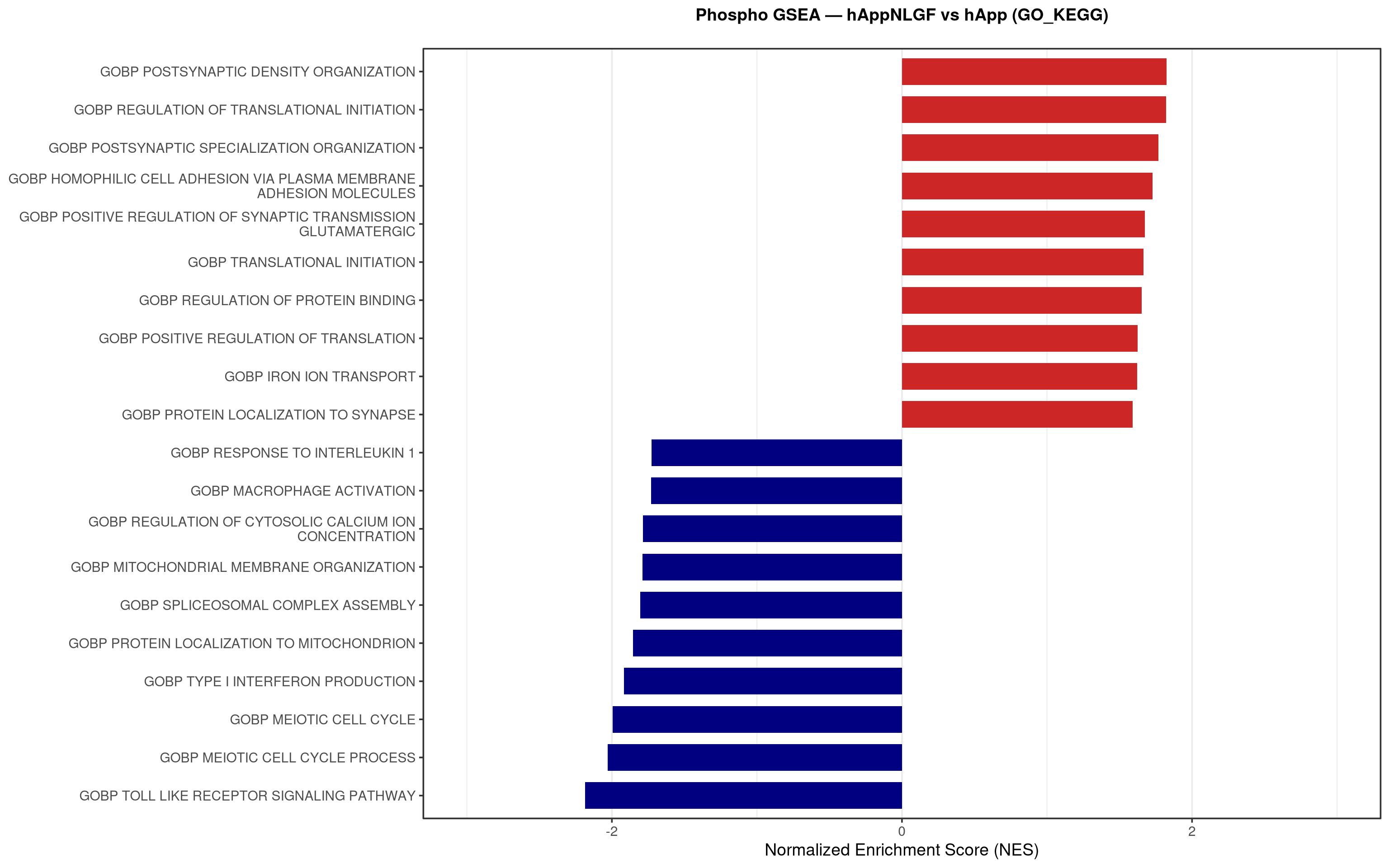
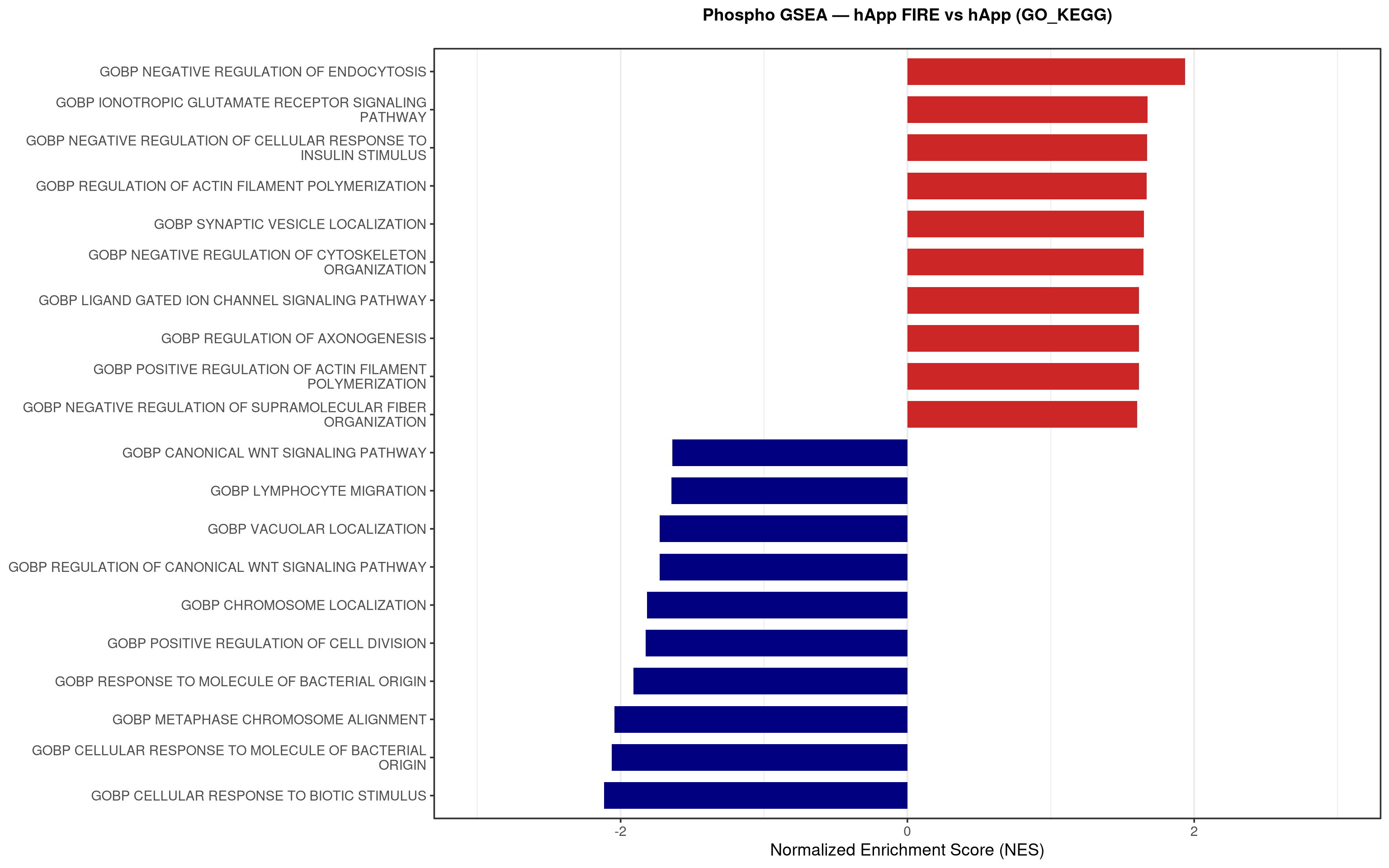
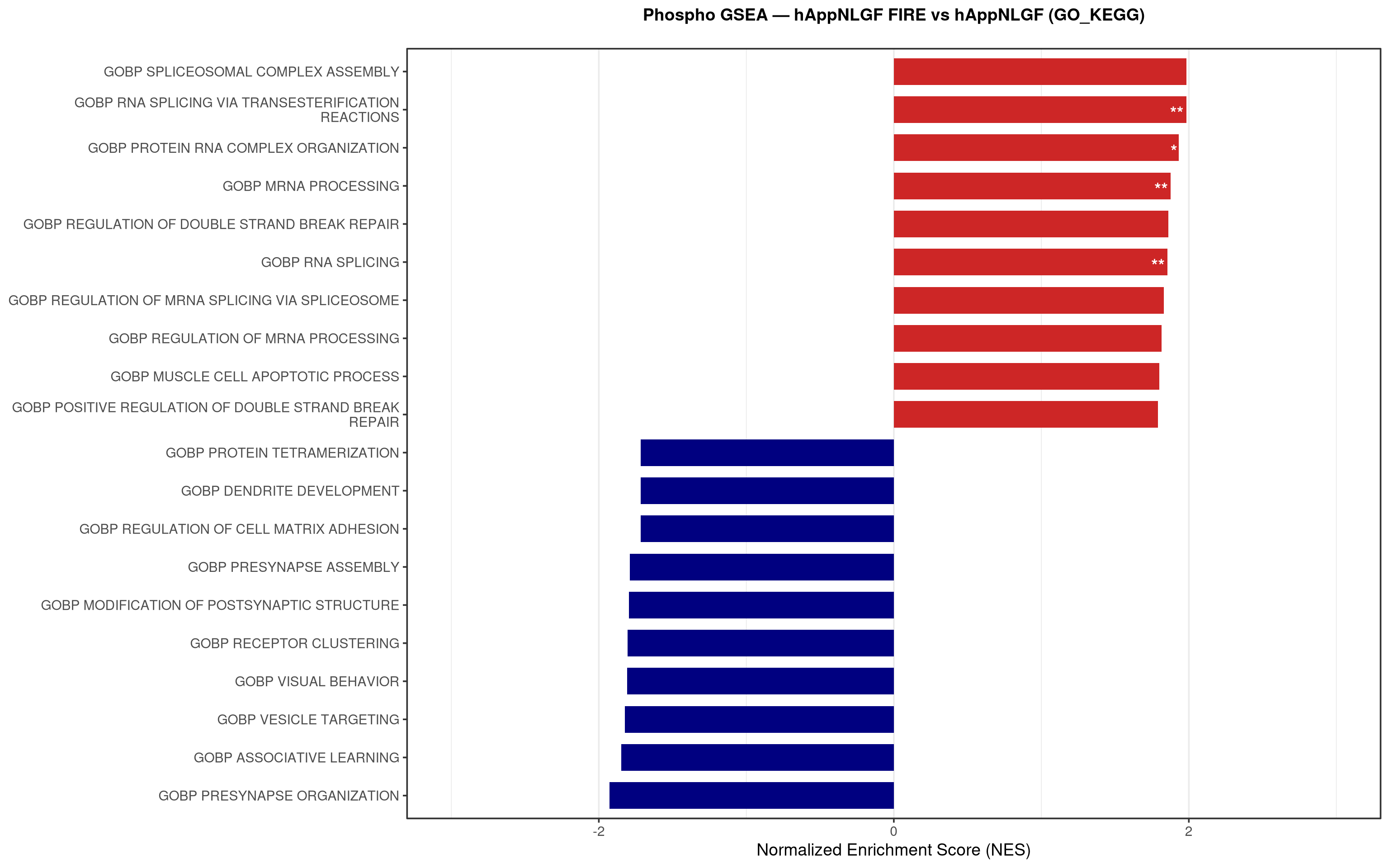
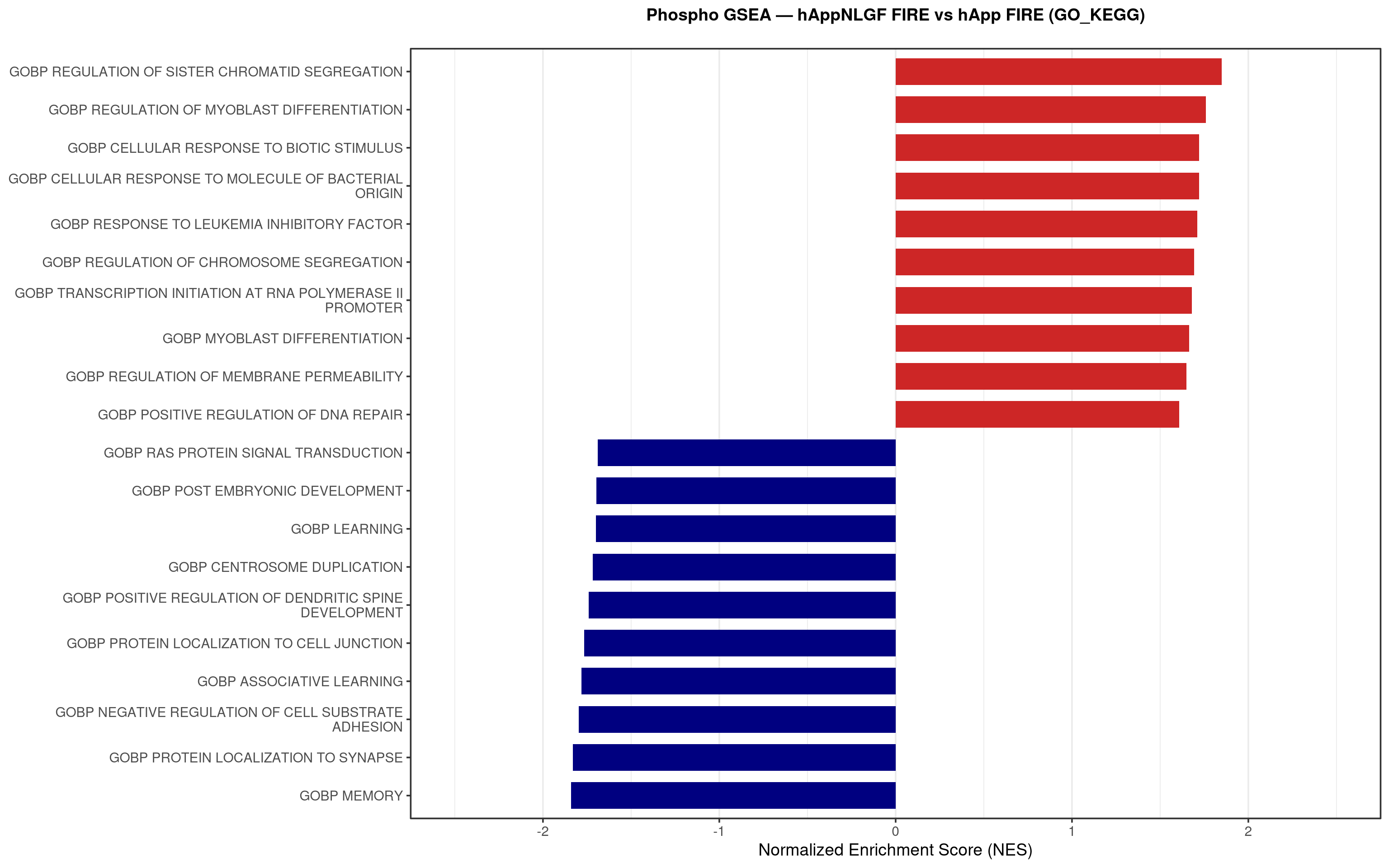
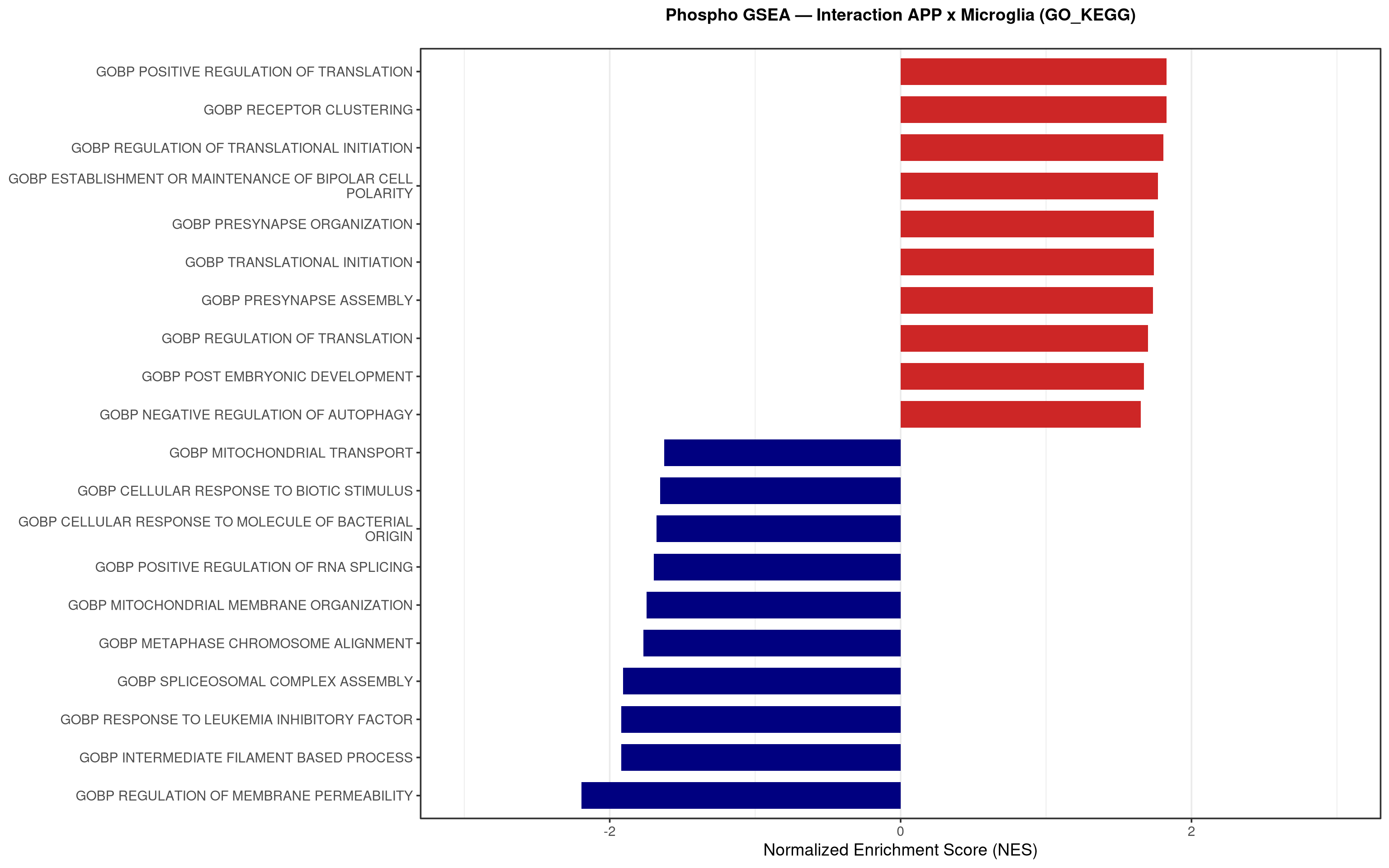
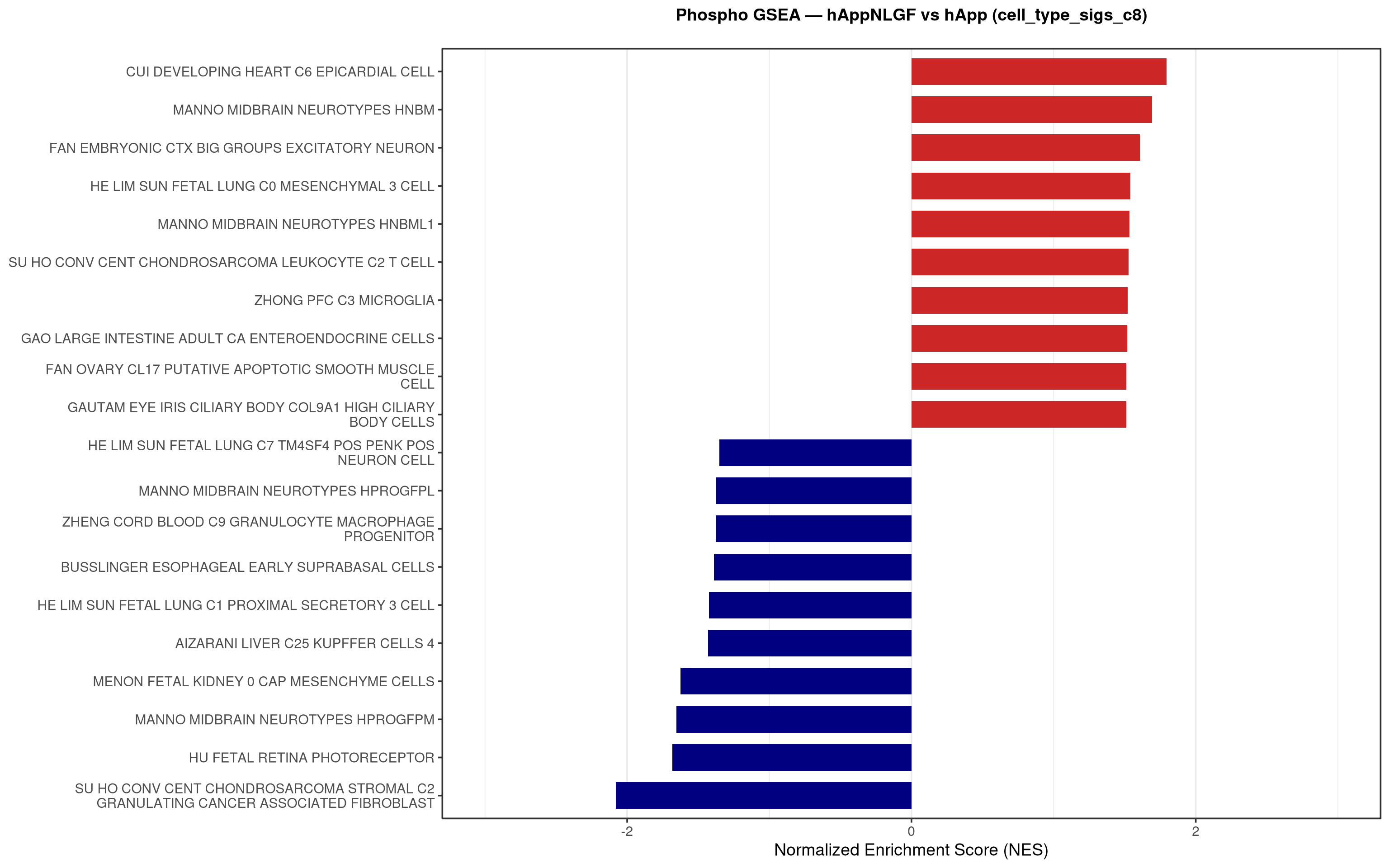
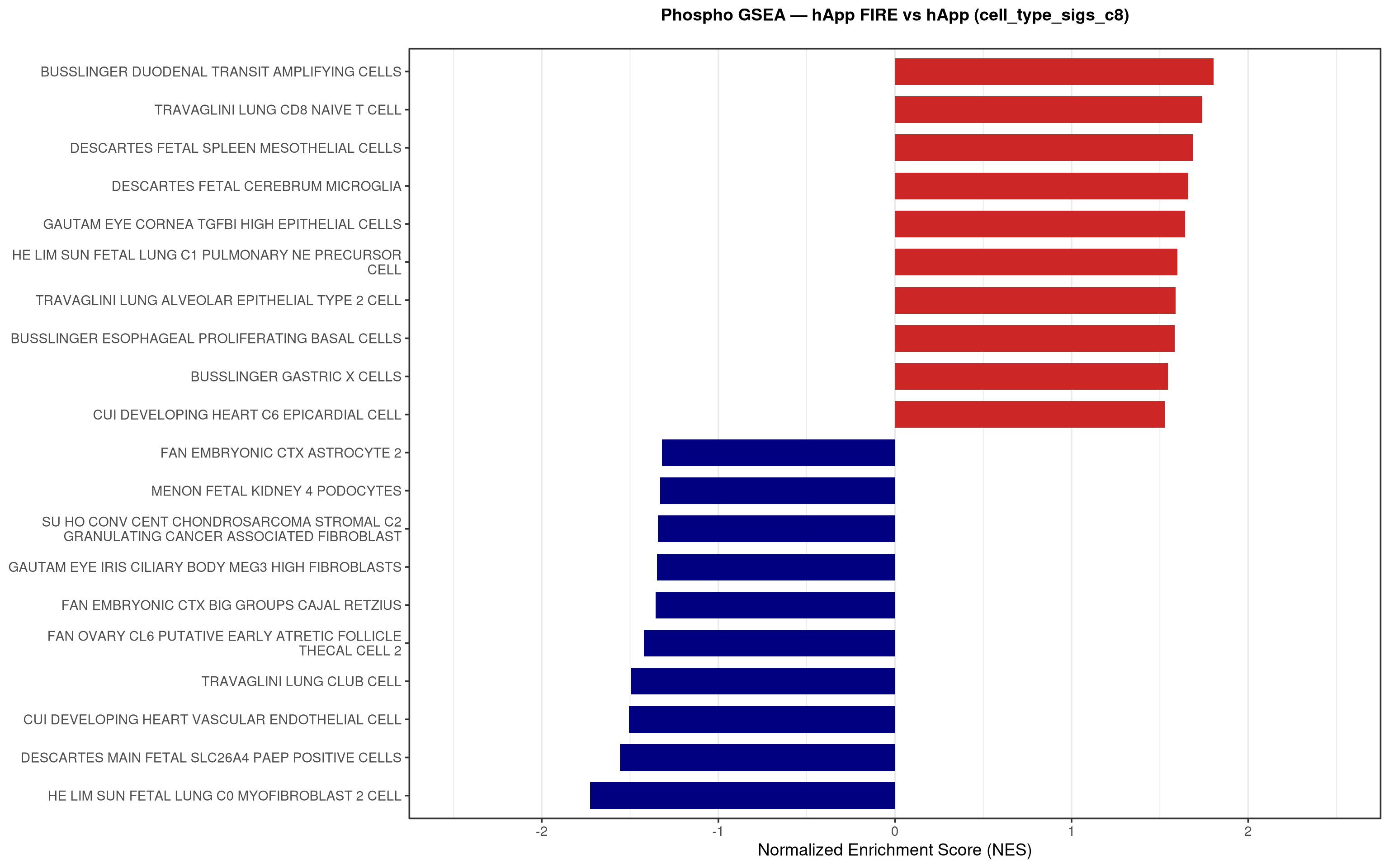
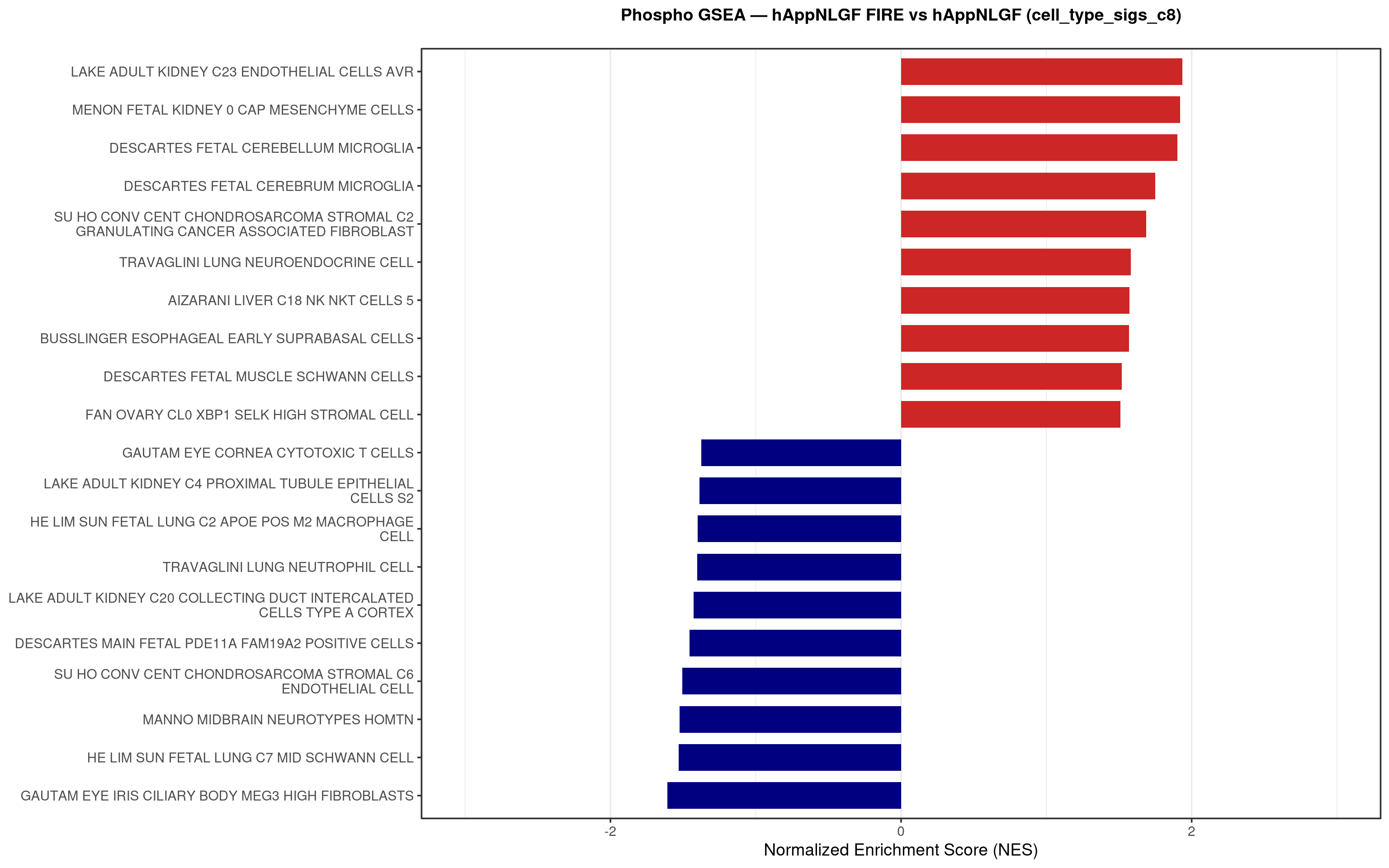
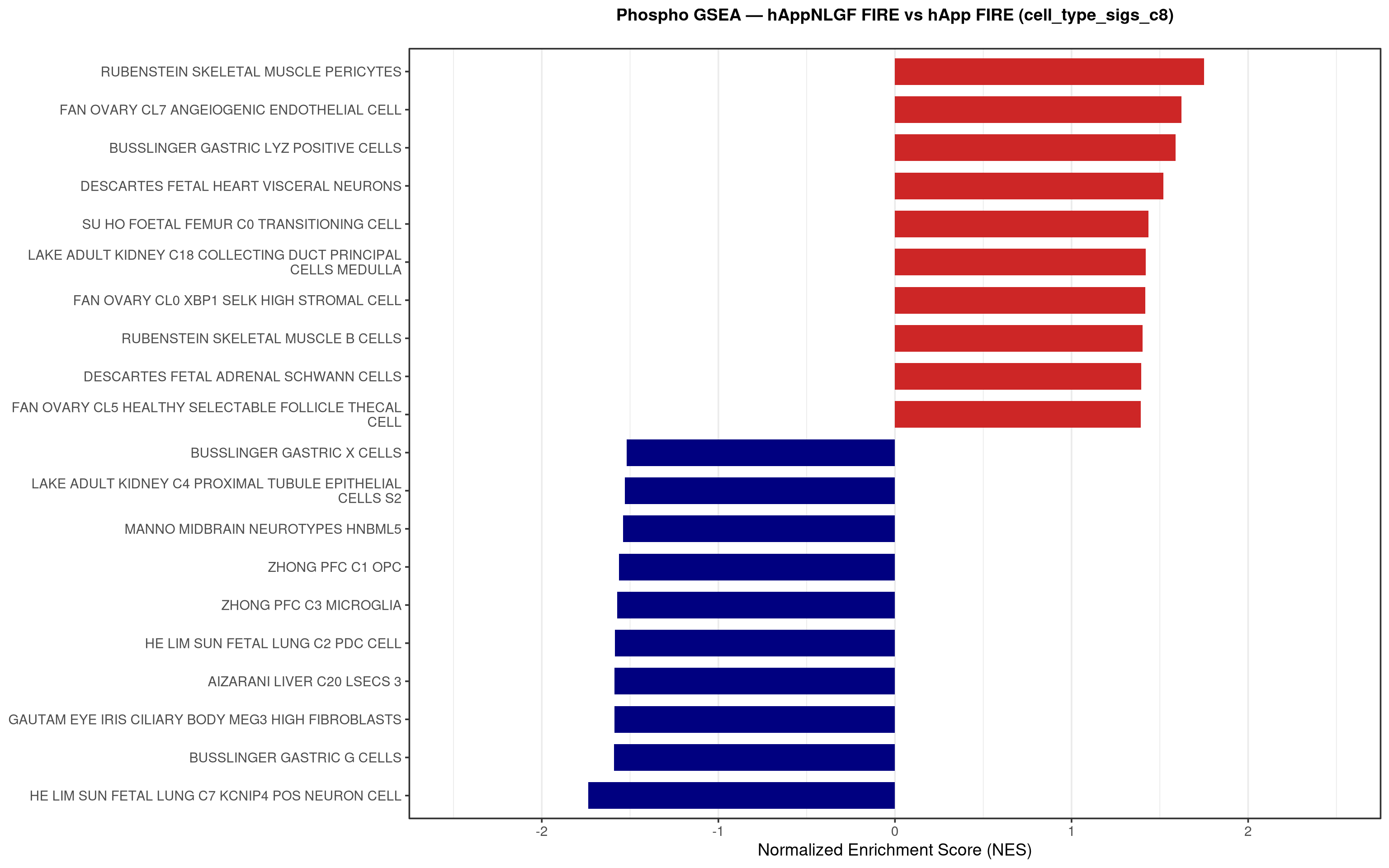
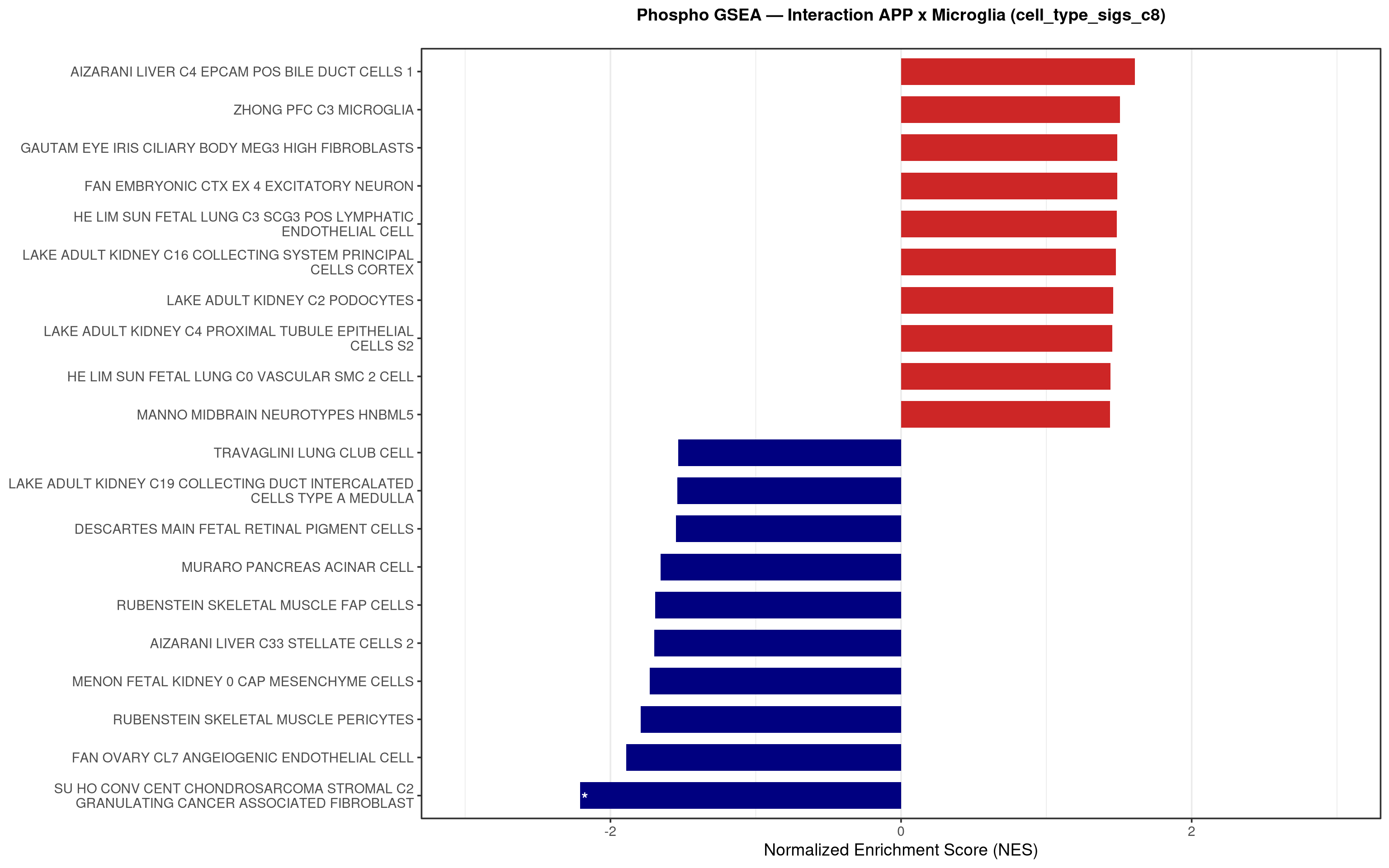
--- title: "5. Phospho — Pathway enrichment" --- ## Overview `bulk_proteomics/notebooks/04_gsea.qmd` .## Libraries ```{r setup, include=FALSE, message=FALSE} library(data.table) library(qs2) library(dplyr) library(stringr) library(glue) library(ggplot2) library(msigdbr) library(fgsea) library(viridis) library(AnnotationDbi) library(org.Mm.eg.db) library(purrr) ``` ## Directories ```{r} <- "/nemo/lab/destrooperb/home/shared/zanettc/giulia_proteomics/phosphoproteomics" <- "run3" <- file.path (base_dir, "results" , run_num)<- file.path (base_dir, "data" , "processed" , run_num)<- file.path (results_dir, "Phospho_GSEA" )dir.create (gsea_dir, recursive = TRUE , showWarnings = FALSE )``` ## Load adjusted phospho results ```{r load} res_df <- fread(file.path(results_dir, "PTM_adjusted_GroupComparison.csv")) res_df[, Accession := str_extract(Protein, "^[^_;]+")] res_df[, Gene := mapIds(org.Mm.eg.db, keys = Accession, column = "SYMBOL", keytype = "UNIPROT", multiVals = "first")] res_df[, Gene := ifelse(is.na(Gene) | Gene == "", Accession, Gene)] ``` ## Build per-contrast gene-level rank vectors ```{r ranks} prep_phospho_ranks <- function(df, contrast_label) { d <- df %>% filter(Label == contrast_label, is.finite(log2FC), !is.na(Gene), Gene != "") %>% mutate(Gene = toupper(Gene)) %>% group_by(Gene) %>% slice_max(order_by = abs(log2FC), n = 1, with_ties = FALSE) %>% ungroup() ranks <- d$log2FC names(ranks) <- d$Gene ranks <- ranks[!is.na(ranks) & !is.na(names(ranks))] sort(ranks, decreasing = TRUE) } contrasts_master <- list( "hAppNLGF_vs_hApp" = prep_phospho_ranks(res_df, "hAppNLGF_vs_hApp"), "hApp_FIRE_vs_hApp" = prep_phospho_ranks(res_df, "hApp_FIRE_vs_hApp"), "hAppNLGF_FIRE_vs_hAppNLGF" = prep_phospho_ranks(res_df, "hAppNLGF_FIRE_vs_hAppNLGF"), "hAppNLGF_FIRE_vs_hApp_FIRE" = prep_phospho_ranks(res_df, "hAppNLGF_FIRE_vs_hApp_FIRE"), "Interaction_APP_x_Microglia" = prep_phospho_ranks(res_df, "Interaction_APP_x_Microglia") ) ``` ## Load mouse gene sets ```{r genesets} go_df <- msigdbr(species = "Mus musculus", collection = "C5", subcollection = "GO:BP") kegg_df <- msigdbr(species = "Mus musculus", collection = "C2", subcollection = "CP:KEGG_MEDICUS") c8_df <- msigdbr(species = "Mus musculus", collection = "C8") go_list <- split(go_df$gene_symbol, go_df$gs_name) kegg_list <- split(kegg_df$gene_symbol, kegg_df$gs_name) c8_list <- split(c8_df$gene_symbol, c8_df$gs_name) gene_sets_master <- list( "GO_KEGG" = c(go_list, kegg_list), "cell_type_sigs_c8" = c8_list ) ``` ## fgsea + barplots (mirrors bulk notebook 04) ```{r run_gsea, message=FALSE, warning=FALSE, fig.width=16, fig.height=10} run_gsea <- function(ranks, comp_name, geneset_name, pathways) { pathways_upper <- lapply(pathways, toupper) set.seed(42) fgsea_res <- fgsea(pathways = pathways_upper, stats = ranks, minSize = 15, maxSize = 3000, nPermSimple = 10000, BPPARAM = BiocParallel::MulticoreParam(workers = 4, progressbar = FALSE)) if (nrow(fgsea_res) == 0) return(NULL) out <- fgsea_res %>% as_tibble() %>% mutate(leadingEdge = sapply(leadingEdge, paste, collapse = ",")) %>% arrange(padj) out_dir <- file.path(gsea_dir, comp_name) dir.create(out_dir, recursive = TRUE, showWarnings = FALSE) write.csv(out, file.path(out_dir, glue("Phospho_GSEA_{geneset_name}.csv")), row.names = FALSE) top <- out %>% arrange(desc(NES)) %>% slice_head(n = 10) %>% bind_rows(out %>% arrange(NES) %>% slice_head(n = 10)) %>% distinct(pathway, .keep_all = TRUE) %>% mutate(sig_label = case_when(padj < 0.001 ~ "***", padj < 0.01 ~ "**", padj < 0.05 ~ "*", TRUE ~ ""), pathway = factor(pathway, levels = unique(pathway[order(NES)]))) if (nrow(top) == 0) return(out) axis_zoom <- max(c(2.5, ceiling(max(abs(top$NES)) * 1.05))) comp_parts <- str_split(comp_name, "_vs_")[[1]] if (length(comp_parts) == 2) { label_A <- glue("{comp_parts[1]} Enriched") label_B <- glue("{comp_parts[2]} Enriched") } else { label_A <- "Numerator Enriched" label_B <- "Denominator Enriched" } p <- ggplot(top, aes(x = NES, y = pathway, fill = ifelse(NES > 0, label_A, label_B))) + geom_col(width = 0.7) + geom_text(aes(label = sig_label, hjust = ifelse(NES > 0, 1.2, -0.2)), color = "white", size = 5, vjust = 0.75, fontface = "bold") + scale_fill_manual(values = setNames(c("firebrick3", "navy"), c(label_A, label_B))) + labs(x = "Normalized Enrichment Score (NES)", y = NULL, title = glue("Phospho GSEA — {gsub('_', ' ', comp_name)} ({geneset_name})")) + theme_bw(base_size = 14) + theme(legend.position = "none", plot.title = element_text(hjust = 0.5, face = "bold", size = 14, margin = margin(b = 20)), panel.grid.major.y = element_blank()) + scale_y_discrete(labels = function(x) str_wrap(gsub("_", " ", x), width = 50)) + coord_cartesian(xlim = c(-axis_zoom, axis_zoom), clip = "off") ggsave(file.path(out_dir, glue("Phospho_GSEA_Barplot_{geneset_name}.pdf")), p, width = 9, height = 7) print(p) out } walk2(names(gene_sets_master), gene_sets_master, function(gset_name, pathways) { walk2(contrasts_master, names(contrasts_master), function(ranks, comp_name) { run_gsea(ranks, comp_name, gset_name, pathways) gc() }) }) ```