Code
library(data.table)
library(qs2)
library(ggrepel)
library(ggplot2)
library(stringr)
library(glue)
library(dplyr)
library(msigdbr)
library(fgsea)
library(viridis)
library(readxl)==================================================================
This script performs GSEA on the MSstats differential-abundance results for the Giulia mouse microglia proteome (hApp / hAppNLGF / hApp_FIRE / hAppNLGF_FIRE). It uses the fgsea package.
1. Data Loading: Imports the MSstats group-comparison table (CSV) from notebook 02a.
2. Gene Set Prep: Loads mouse pathways from msigdbr (GO / KEGG / cell-type C8).
3. Ranking: Ranks proteins by Log2FC for every contrast.
4. Analysis Loop: Runs GSEA for every combination of [Comparison x Gene Set Database].
5. Visualization:
==================================================================
library(data.table)
library(qs2)
library(ggrepel)
library(ggplot2)
library(stringr)
library(glue)
library(dplyr)
library(msigdbr)
library(fgsea)
library(viridis)
library(readxl)base_dir <- "/nemo/lab/destrooperb/home/shared/zanettc/giulia_proteomics/bulk_proteomics"
run_num <- "run4"
results_dir <- file.path(base_dir, "results", run_num)
objects_dir <- file.path(base_dir, "data", "processed", run_num)
input_dir <- file.path(base_dir, "input")
dir.create(results_dir, recursive = TRUE, showWarnings = FALSE)
dir.create(objects_dir, recursive = TRUE, showWarnings = FALSE)#seurat <- qs_read(glue("output/{run_num}/objects/prelabelled_integrated_rpca.qs2"))
res_df <- fread(file.path(results_dir, "Full_GroupComparison_Results.csv"))
clean_spec_raw <- fread(file.path(base_dir, "data/processed/run1/clean_spec_raw.csv"))
protein_dictionary <- fread(file.path(objects_dir, 'protein_dictionary.csv'))# ==============================================================================
# 1. HELPER FUNCTIONS
# ==============================================================================
# A. Create Rank Vector from MSstats Data
prep_msstats_ranks <- function(msstats_res, dict, comp_label) {
df <- msstats_res %>%
filter(Label == comp_label) %>%
# Keep only finite log2FC values (removes Inf and -Inf)
filter(is.finite(log2FC)) %>%
left_join(dict, by = "Protein") %>%
filter(!is.na(Gene) & Gene != "") %>%
mutate(Gene = toupper(Gene)) %>%
mutate(adj.pvalue = ifelse(adj.pvalue == 0, 1e-300, adj.pvalue)) %>%
group_by(Gene) %>%
slice_max(order_by = abs(log2FC), n = 1, with_ties = FALSE) %>%
ungroup()
#rank by logFC
ranks <- df$log2FC
names(ranks) <- df$Gene
# Remove any unexpected NAs in names or values
ranks <- ranks[!is.na(ranks) & !is.na(names(ranks))]
return(sort(ranks, decreasing = TRUE))
}
# B. Core GSEA & Plotting Function
run_gsea_analysis <- function(ranks,
comparison_name,
geneset_name,
pathways,
base_objects_dir,
base_graphs_dir,
color_A = "firebrick3",
color_B = "navy") {
message(glue("Running GSEA | Set: {geneset_name} | Comp: {comparison_name}"))
# --- DYNAMIC LABEL GENERATION ---
# Split the comparison name to get the two groups (e.g., "PlaqueNear" and "PlaqueFar")
comp_parts <- str_split(comparison_name, "_vs_")[[1]]
if(length(comp_parts) == 2) {
group_A_label <- glue("{comp_parts[1]} Enriched") # Positive NES
group_B_label <- glue("{comp_parts[2]} Enriched") # Negative NES
} else {
group_A_label <- "Group A Enriched"
group_B_label <- "Group B Enriched"
}
# Clean title for the plot
clean_comp_title <- gsub("_", " ", comparison_name)
plot_title <- glue("{clean_comp_title} ({geneset_name})")
# --- Directories ---
current_objects_dir <- file.path(base_objects_dir, "GSEA_Analysis", comparison_name)
current_graphs_dir <- file.path(base_graphs_dir, "GSEA_Analysis", comparison_name)
dir.create(current_objects_dir, recursive = TRUE, showWarnings = FALSE)
dir.create(current_graphs_dir, recursive = TRUE, showWarnings = FALSE)
# --- Run FGSEA ---
pathways_upper <- lapply(pathways, toupper)
set.seed(42)
fgsea_res <- fgsea(
pathways = pathways_upper,
stats = ranks,
minSize = 15,
maxSize = 3000,
nPermSimple = 10000,
BPPARAM = BiocParallel::MulticoreParam(workers = 4, progressbar = FALSE)
)
if (nrow(fgsea_res) == 0) {
warning(glue("No significant pathways for {geneset_name} in {comparison_name}"))
return(NULL)
}
# --- Tidy & Save ---
fgsea_res_tidy <- fgsea_res %>%
as_tibble() %>%
mutate(leadingEdge = sapply(leadingEdge, paste, collapse = ",")) %>%
mutate(pathway = gsub("_geneset", "", pathway)) %>%
arrange(padj)
write.csv(fgsea_res_tidy, file.path(current_objects_dir, glue("Table_{geneset_name}.csv")), row.names = FALSE)
# --- Barplot ---
top_gsea <- fgsea_res_tidy %>%
arrange(desc(NES)) %>%
slice_head(n = 10) %>%
bind_rows(fgsea_res_tidy %>% arrange(NES) %>% slice_head(n = 10)) %>%
distinct(pathway, .keep_all = TRUE) %>%
mutate(
sig_label = case_when(
padj < 0.001 ~ "***",
padj < 0.01 ~ "**",
padj < 0.05 ~ "*",
TRUE ~ ""
),
# Apply dynamic labels based on NES direction
fill_group = ifelse(NES > 0, group_A_label, group_B_label),
pathway = factor(pathway, levels = unique(pathway[order(NES)]))
)
if (nrow(top_gsea) == 0) return(fgsea_res_tidy)
axis_zoom <- 3.5
p_bar <- ggplot(top_gsea, aes(x = NES, y = pathway, fill = fill_group)) +
geom_col(width = 0.7) +
geom_text(aes(label = sig_label, hjust = ifelse(NES > 0, 1.2, -0.2)),
color = "white", size = 5, vjust = 0.75, fontface = "bold") +
scale_fill_manual(values = setNames(c(color_A, color_B), c(group_A_label, group_B_label))) +
scale_x_continuous(breaks = seq(-axis_zoom, axis_zoom, 0.5),
labels = function(x) ifelse(x %% 1 == 0, x, "")) +
labs(x = "Normalized Enrichment Score (NES)", y = NULL, title = plot_title) +
theme_bw(base_size = 14) +
theme(
panel.grid.major.x = element_line(color = "grey92", linewidth = 0.5),
panel.grid.major.y = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_rect(colour = "black", fill = NA, linewidth = 1.5),
legend.position = "none",
plot.title = element_text(hjust = 0.5, face = "bold", size = 16, margin = margin(b = 40)),
plot.margin = margin(t = 40, r = 20, b = 20, l = 20, unit = "pt"),
#ensure y axis has enough room, and tightens lines together
axis.text.y = element_text(size = 9, lineheight = 0.8)
) +
scale_y_discrete(labels = function(x) str_wrap(gsub("_", " ", x), width = 50)) +
# Dynamic text annotations at the top of the plot
annotate("text", x = -axis_zoom/2, y = Inf,
label = glue("^ {group_B_label}"), color = color_B, size = 5, fontface = "bold", vjust = -1.0) +
annotate("text", x = axis_zoom/2, y = Inf,
label = glue("^ {group_A_label}"), color = color_A, size = 5, fontface = "bold", vjust = -1.0) +
coord_cartesian(xlim = c(-axis_zoom, axis_zoom), clip = "off")
dyn_h <- 2 + (nrow(top_gsea) * 0.3)
dyn_w <- 5 + (max(nchar(as.character(top_gsea$pathway))) * 0.12)
print(p_bar)
ggsave(filename = file.path(current_graphs_dir, glue("Barplot_{geneset_name}.pdf")),
plot = p_bar, width = dyn_w, height = dyn_h, limitsize = FALSE)
# --- Dotplot (Top 10 Highest NES) ---
top_nes_gsea <- fgsea_res_tidy %>%
arrange(desc(NES)) %>%
slice_head(n = 10) %>%
mutate(pathway = factor(pathway, levels = unique(pathway[order(NES)])))
if (nrow(top_nes_gsea) > 0) {
# Define our wrap width
wrap_width <- 45
p_sig <- ggplot(top_nes_gsea, aes(x = NES, y = pathway, color = NES, size = -log10(padj))) +
geom_vline(xintercept = 0, linetype = "dashed", color = "grey60") +
geom_point(alpha = 0.8) +
scale_color_gradient2(low = color_B, mid = "grey90", high = color_A, midpoint = 0, name = "NES") +
scale_size_continuous(range = c(3, 8), name = "-log10(padj)") +
labs(x = "Normalized Enrichment Score (NES)", y = NULL,
title = glue("Top 10 Highest NES:\n{clean_comp_title} ({geneset_name})")) +
theme_bw(base_size = 14) +
theme(
panel.grid.major.x = element_line(color = "grey92", linewidth = 0.5),
panel.grid.major.y = element_line(color = "grey92", linewidth = 0.5, linetype = "dotted"),
panel.grid.minor = element_blank(),
panel.border = element_rect(colour = "black", fill = NA, linewidth = 1.5),
plot.title = element_text(hjust = 0.5, face = "bold", size = 14, margin = margin(b = 20)),
plot.margin = margin(t = 20, r = 10, b = 10, l = 10, unit = "pt"),
axis.text.y = element_text(lineheight = 0.8)
) +
scale_y_discrete(labels = function(x) str_wrap(gsub("_", " ", x), width = wrap_width))
# --- DYNAMIC SIZING ---
# Cap the character count at wrap_width so the plot doesn't stretch endlessly horizontally
max_chars <- min(max(nchar(as.character(top_nes_gsea$pathway))), wrap_width)
# Calculate dimensions: enough base room for the plot/legends + scaled room for text/points
dyn_h_sig <- 3 + (nrow(top_nes_gsea) * 0.4)
dyn_w_sig <- 4.5 + (max_chars * 0.1)
ggsave(filename = file.path(current_graphs_dir, glue("Dotplot_Top10_NES_{geneset_name}.pdf")),
plot = p_sig, width = dyn_w_sig, height = dyn_h_sig, limitsize = FALSE)
}
return(fgsea_res_tidy)
}# ==============================================================================
# 2. CONFIG & DATA LOADING
# ==============================================================================
GLOBAL_CONFIG <- list(
output_dir = file.path(results_dir, "GSEA_Results_Master"),
group_A = "",
group_B = "",
col_A = "firebrick3",
col_B = "navy"
)
# Load Gene Sets (Mus musculus)
#GO (C5: GOBP/GOCC/GOMF) + KEGG (C2: CP:KEGG_MEDICUS)
go_df <- msigdbr(species = "Mus musculus", collection = "C5", subcollection = "GO:BP")
go_list <- split(x = go_df$gene_symbol, f = go_df$gs_name)
kegg_df <- msigdbr(species = "Mus musculus", collection = "C2", subcollection = "CP:KEGG_MEDICUS")
kegg_list <- split(x = kegg_df$gene_symbol, f = kegg_df$gs_name)
go_kegg_list <- c(go_list, kegg_list)
#cell type signature sets
c8_df <- msigdbr(species = "Mus musculus", collection = "C8")
c8_list <- split(x = c8_df$gene_symbol, f = c8_df$gs_name)
# Create master list
gene_sets_master <- list(
"GO_KEGG" = go_kegg_list,
"cell_type_sigs_c8" = c8_list
)
#Create Rank Vectors directly from MSstats output
comparisons_master <- list(
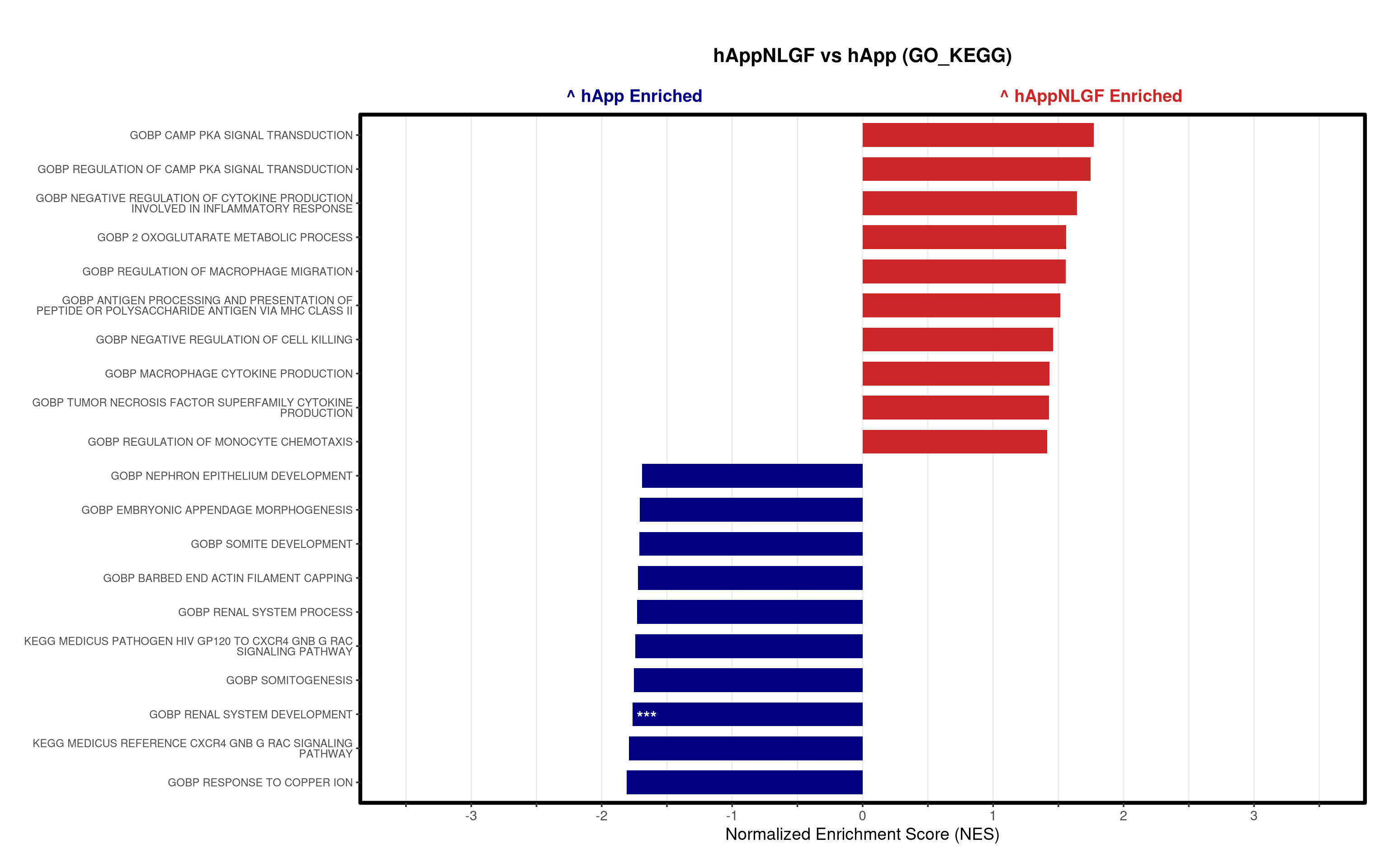
"hAppNLGF_vs_hApp" = prep_msstats_ranks(res_df, protein_dictionary, "hAppNLGF_vs_hApp"),
"hApp_FIRE_vs_hApp" = prep_msstats_ranks(res_df, protein_dictionary, "hApp_FIRE_vs_hApp"),
"hAppNLGF_FIRE_vs_hAppNLGF" = prep_msstats_ranks(res_df, protein_dictionary, "hAppNLGF_FIRE_vs_hAppNLGF"),
"hAppNLGF_FIRE_vs_hApp_FIRE" = prep_msstats_ranks(res_df, protein_dictionary, "hAppNLGF_FIRE_vs_hApp_FIRE")
)# ==============================================================================
# 3. EXECUTION LOOP
# ==============================================================================
purrr::walk2(names(gene_sets_master), gene_sets_master, function(gset_name, gset_pathways) {
purrr::walk2(comparisons_master, names(comparisons_master), function(ranks, comp_name) {
run_gsea_analysis(
ranks = ranks,
comparison_name = comp_name,
geneset_name = gset_name,
pathways = gset_pathways,
base_objects_dir = objects_dir,
base_graphs_dir = results_dir,
color_A = GLOBAL_CONFIG$col_A,
color_B = GLOBAL_CONFIG$col_B
)
gc() # Memory cleanup
})
})
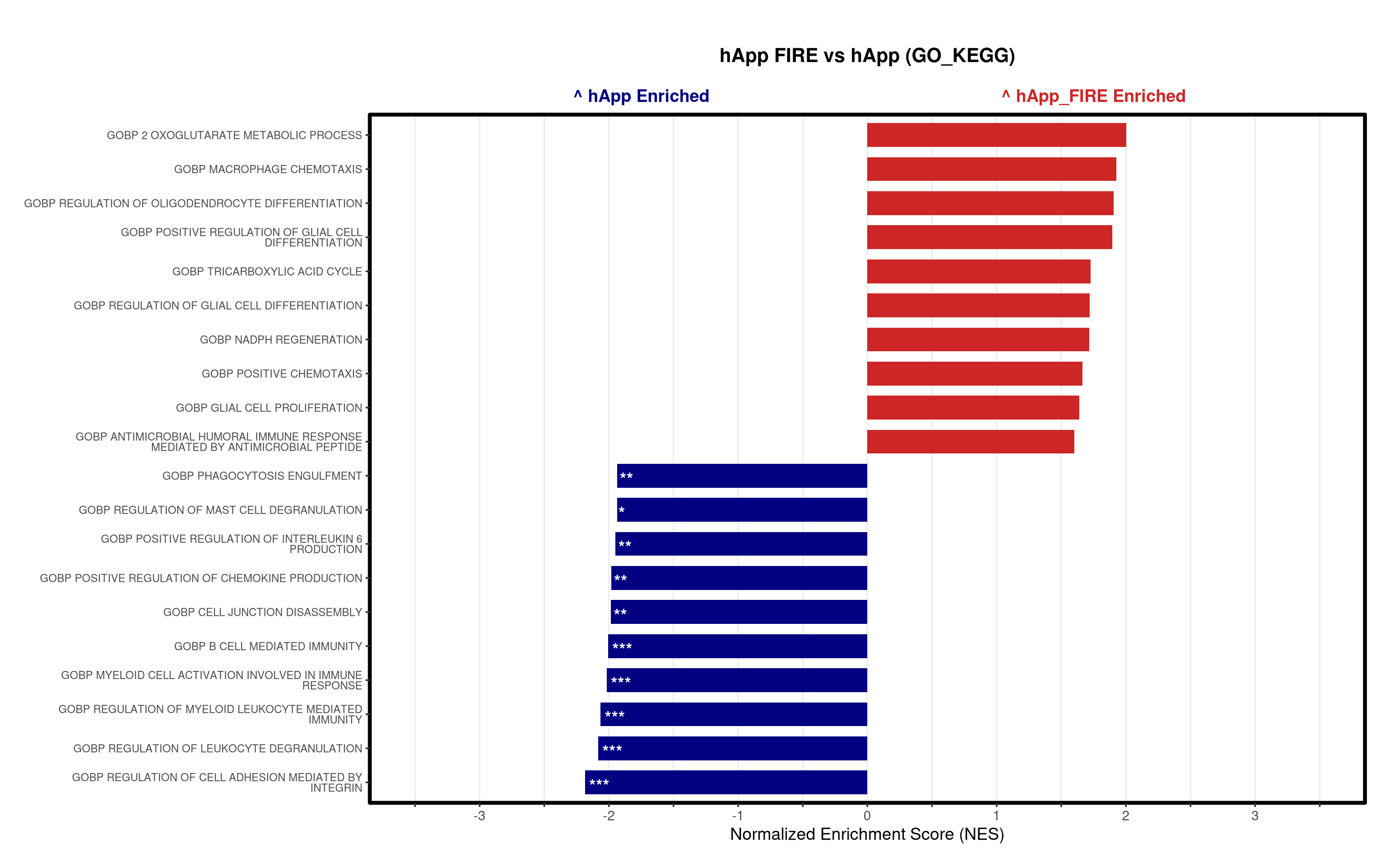
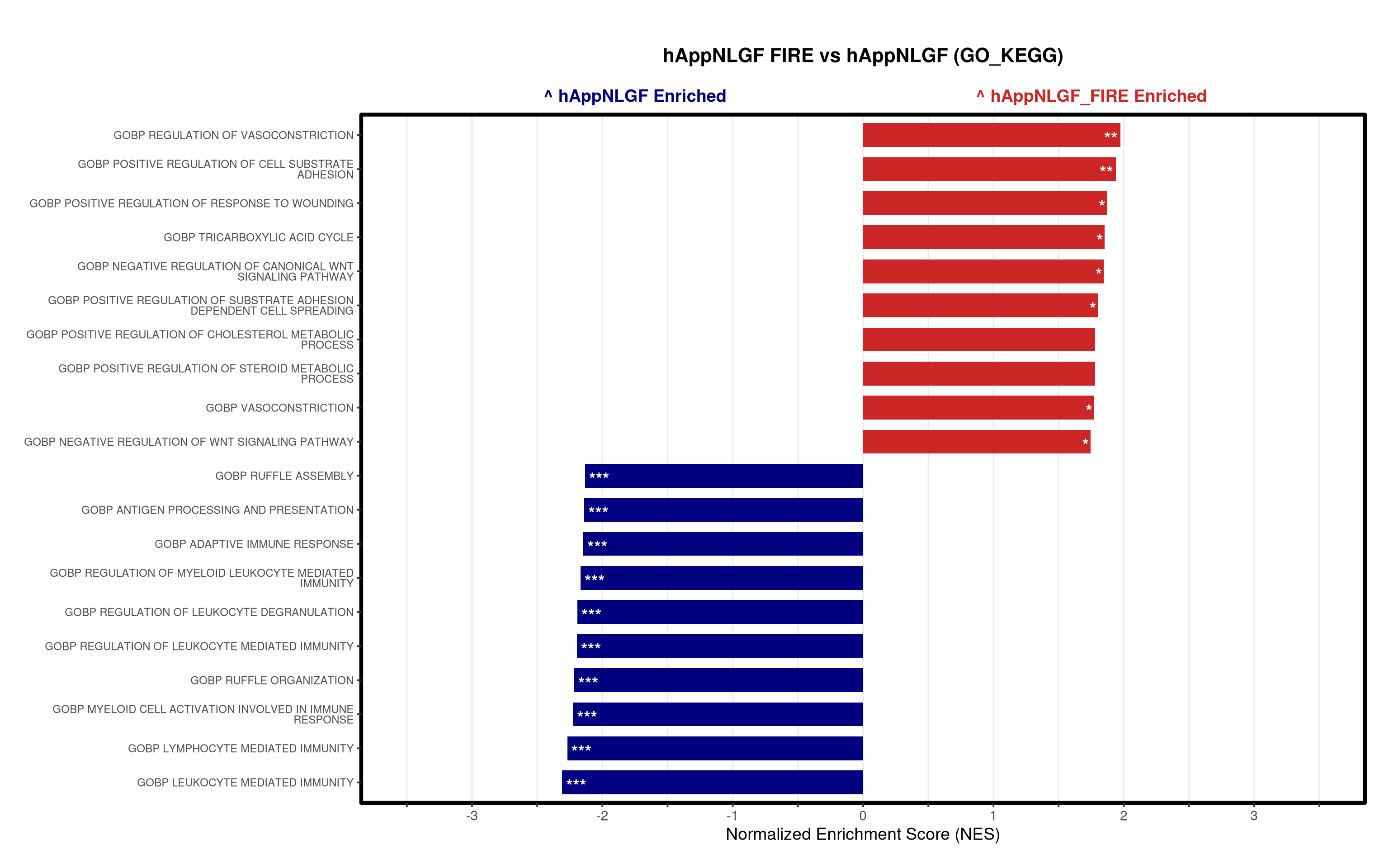
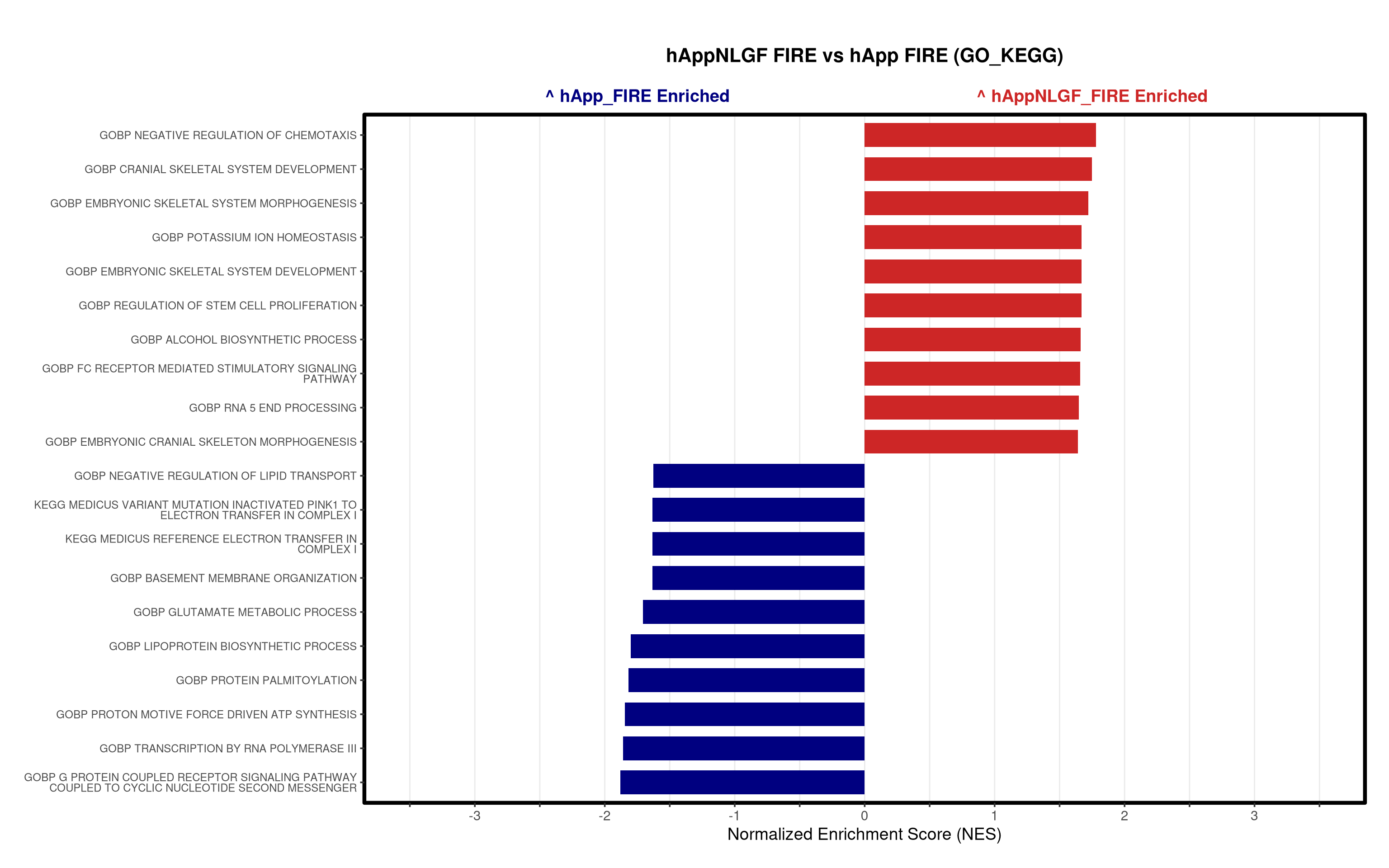
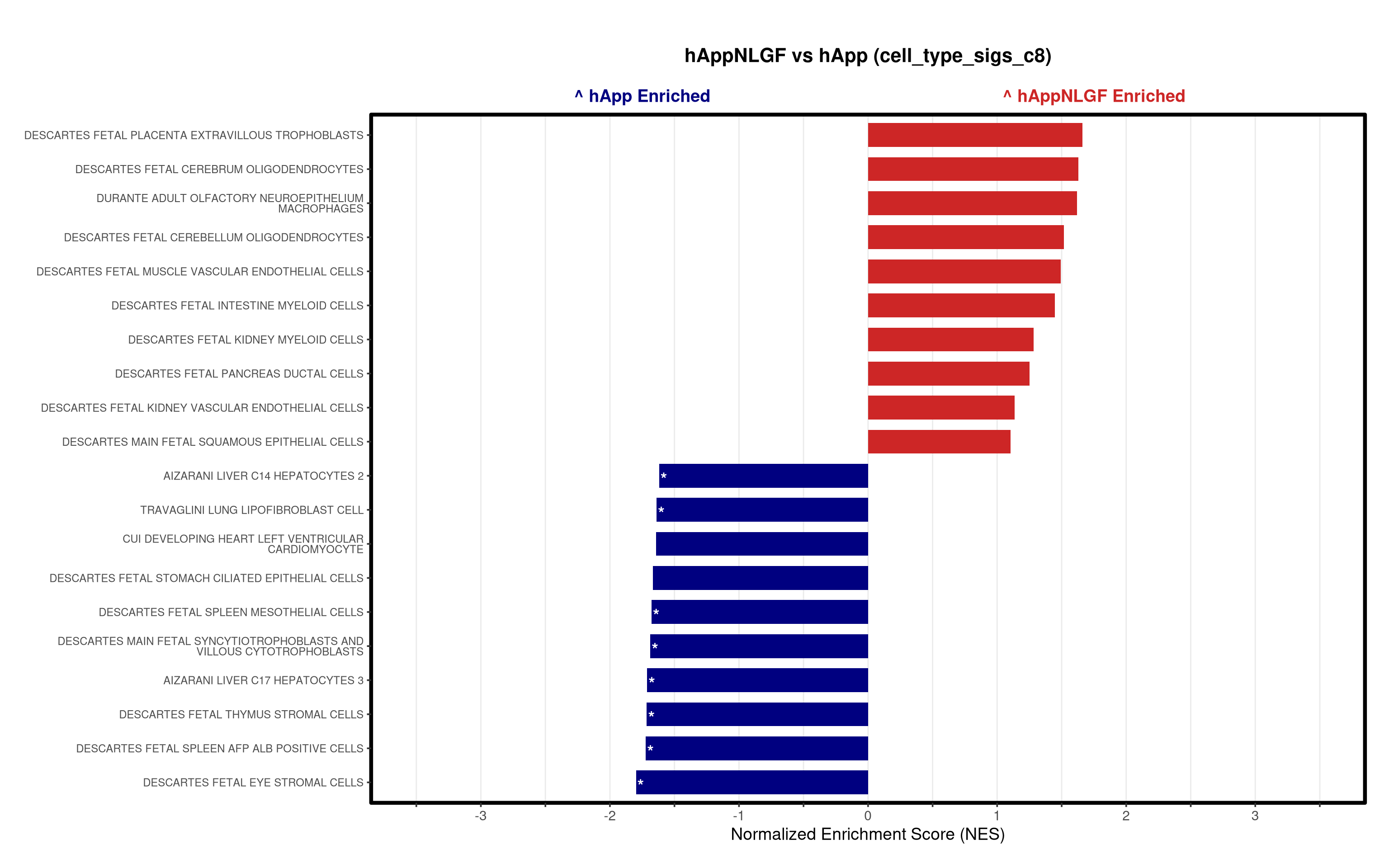
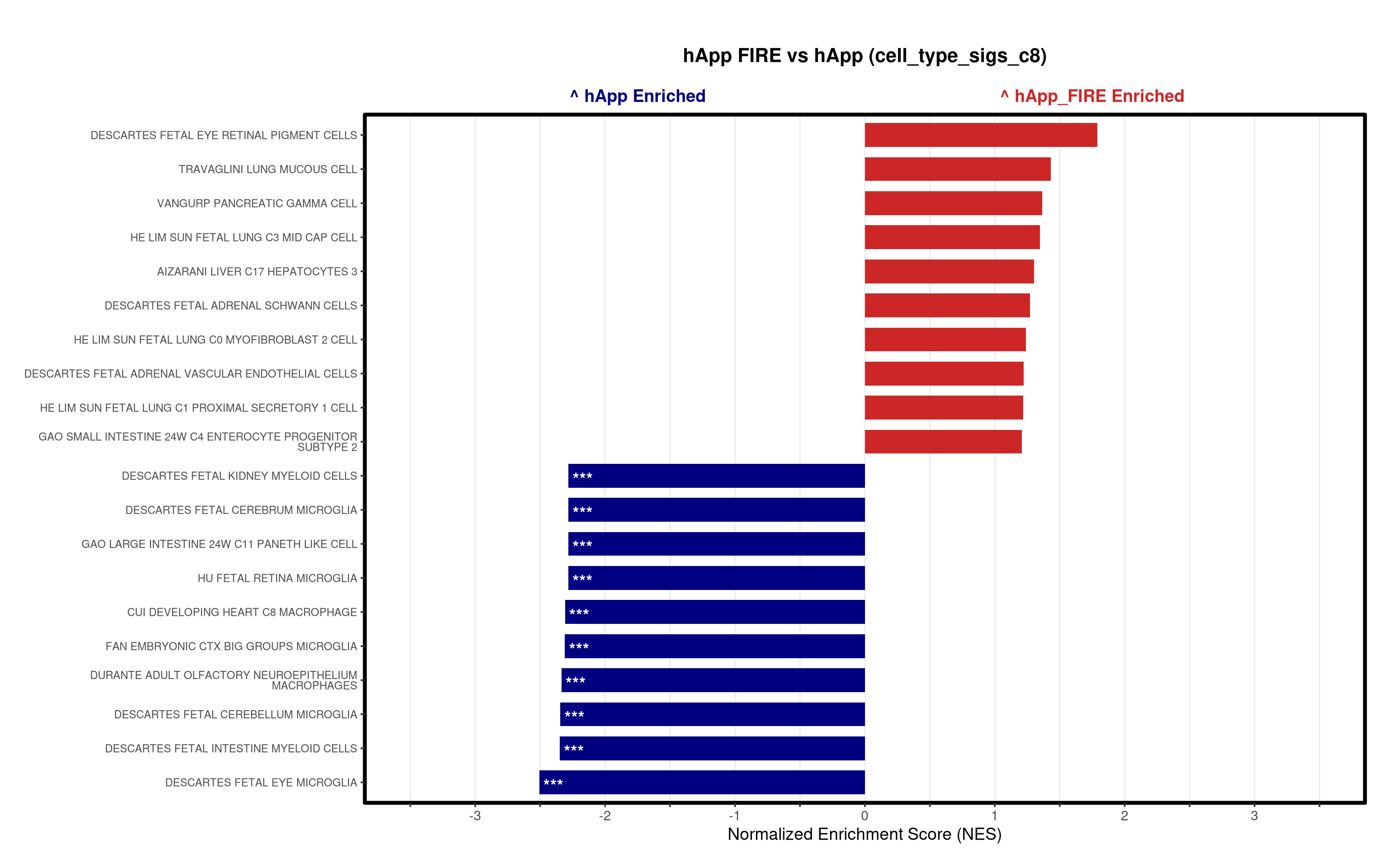
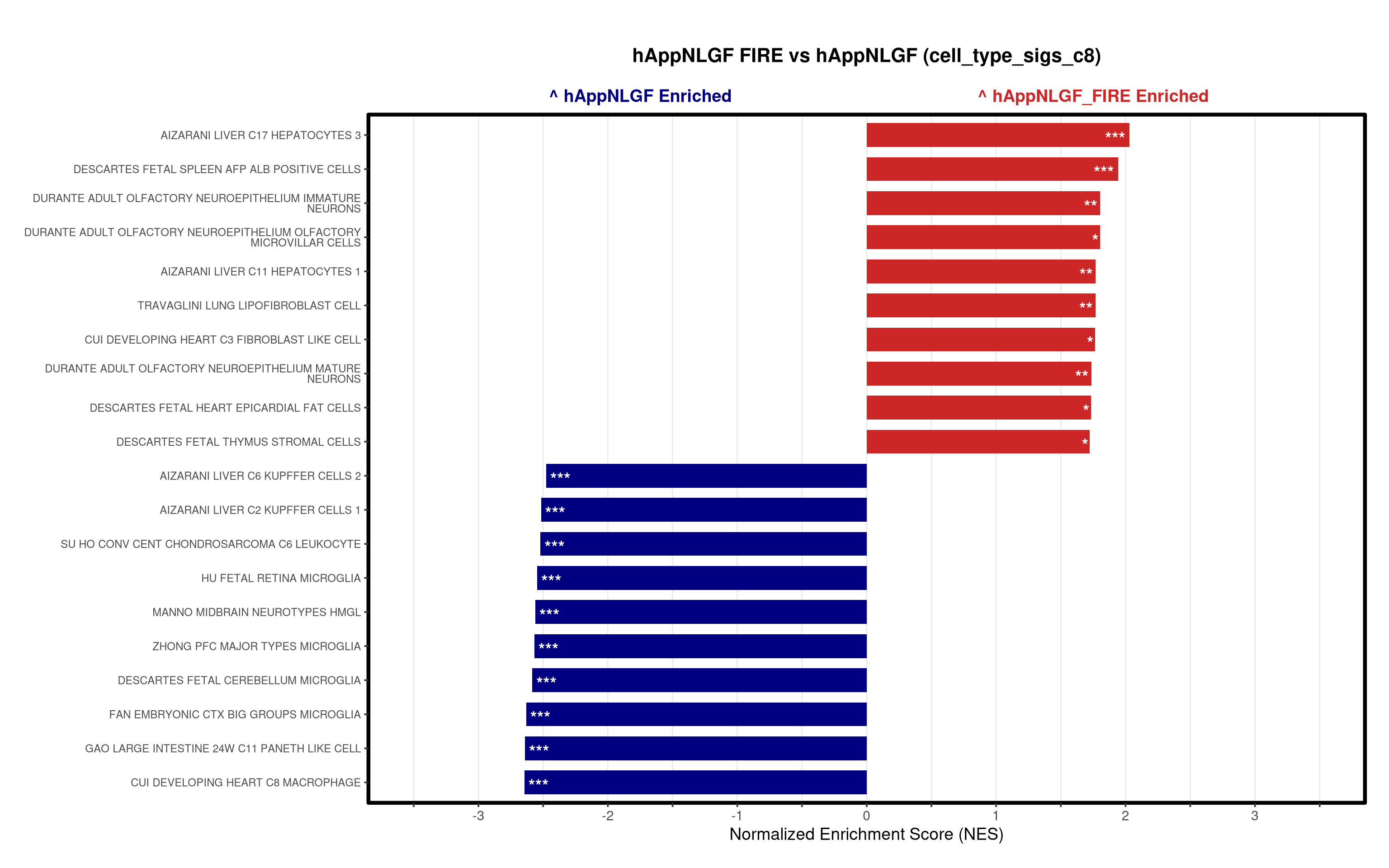
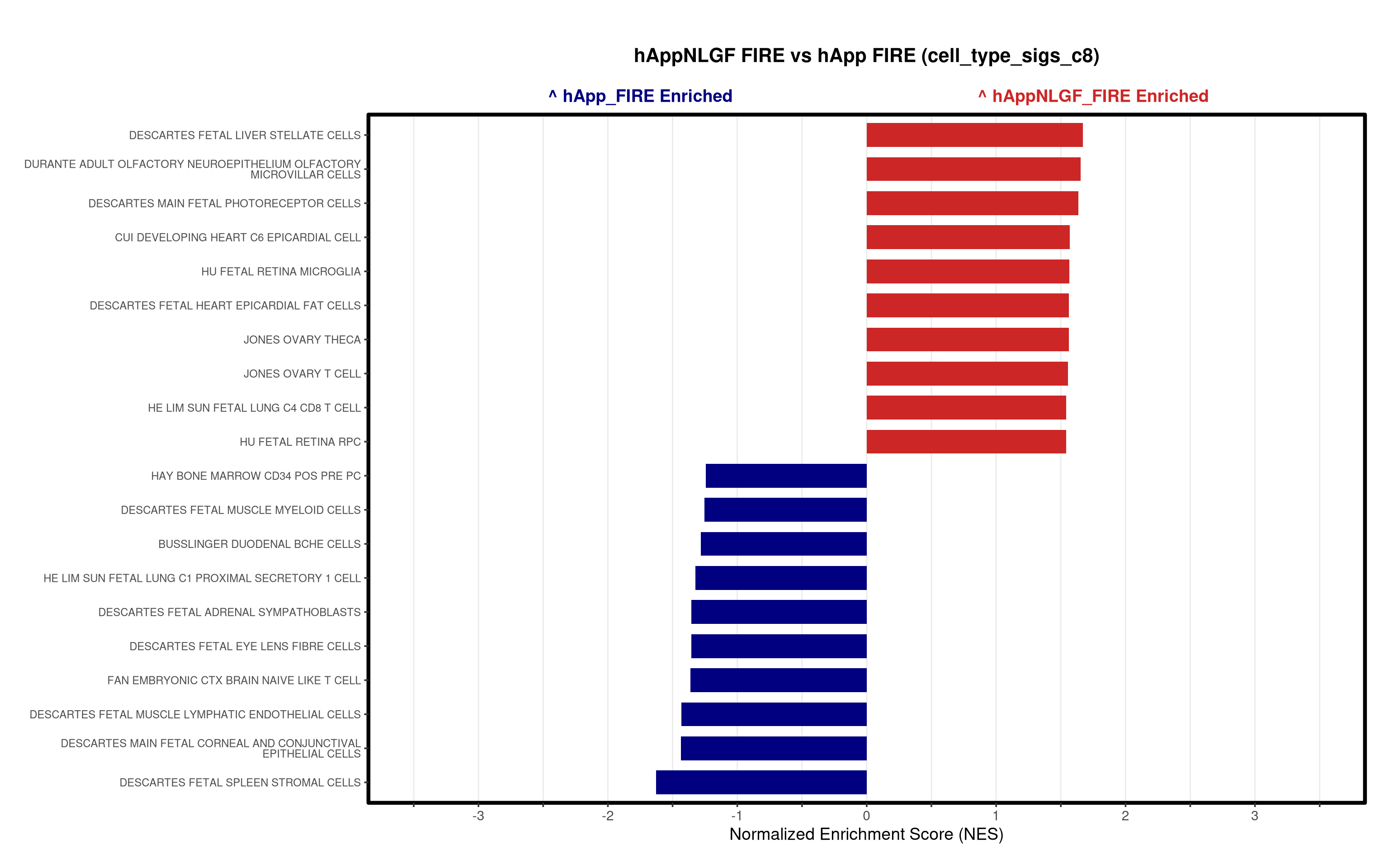
# ==============================================================================
# SPECIFIC TERM PLOTTING
# ==============================================================================
term_to_plot <- "GOBP_PLATELET_AGGREGATION"
# 1. Flatten master gene list
all_pathways_flat <- do.call(c, unname(gene_sets_master))
# Check if the term exists before looping - in case typo
if (!term_to_plot %in% names(all_pathways_flat)) {
stop(glue("Could not find pathway '{term_to_plot}' in gene_sets_master."))
}
pathway_genes <- all_pathways_flat[[term_to_plot]]
# Create a specific output folder
specific_graphs_dir <- file.path(results_dir, "Specific_Pathway_Plots")
dir.create(specific_graphs_dir, showWarnings = FALSE, recursive = TRUE)
# Loop and Plot
purrr::walk2(comparisons_master, names(comparisons_master), function(ranks, comp_name) {
message(glue("Plotting {term_to_plot} for {comp_name}..."))
# Generate the GSEA enrichment plot
gsea_plot <- plotEnrichment(pathway_genes, ranks) +
labs(
title = term_to_plot,
subtitle = glue("Comparison: {comp_name}"),
x = "Rank",
y = "Enrichment Score"
) +
theme_bw(base_size = 14) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5, color = "grey40"),
panel.grid = element_blank()
)
print(gsea_plot)
# Save
ggsave(
filename = file.path(specific_graphs_dir, glue("{term_to_plot}_{comp_name}.pdf")),
plot = gsea_plot,
width = 8,
height = 5
)
})
Tests whether the top 10 up-regulated proteins from a given MSstats contrast overlap the top 10 leading-edge genes of a chosen pathway more than expected by chance. Background = all genes present in that contrast’s rank vector.
target_comparison <- "hAppNLGF_FIRE_vs_hAppNLGF" # change to any of the four contrasts
target_pathway <- term_to_plot # GOBP_LEUKOCYTE_MEDIATED_IMMUNITY by default
# Top 10 up-regulated genes from the chosen contrast
ranks_target <- comparisons_master[[target_comparison]]
top10_contrast <- ranks_target %>%
sort(decreasing = TRUE) %>%
head(10) %>%
names()
# Top 10 leading-edge genes from the chosen pathway in that contrast
pathway_genes_upper <- toupper(all_pathways_flat[[target_pathway]])
set.seed(42)
fgsea_single <- fgsea(
pathways = list(sel = pathway_genes_upper),
stats = ranks_target,
minSize = 15,
maxSize = 3000,
nPermSimple = 10000,
BPPARAM = BiocParallel::MulticoreParam(workers = 4, progressbar = FALSE)
)
leading_edge_genes <- fgsea_single$leadingEdge[[1]]
top10_pathway <- head(leading_edge_genes, 10)
# Background = all ranked genes in this contrast
background_genes <- names(ranks_target)
N <- length(background_genes)
overlap <- intersect(top10_contrast, top10_pathway)
k <- length(overlap) # observed overlap
m <- sum(top10_pathway %in% background_genes) # pathway top10 present in background
n <- N - m # genes NOT in pathway top10
q <- length(top10_contrast) # size of contrast top10
# phyper(k-1, ..., lower.tail = FALSE) gives P(X >= k)
p_val <- phyper(k - 1, m, n, q, lower.tail = FALSE)
cat(glue("=== Hypergeometric Test: {target_comparison} top10 vs {target_pathway} top10 ===\n"))=== Hypergeometric Test: hAppNLGF_FIRE_vs_hAppNLGF top10 vs GOBP_PLATELET_AGGREGATION top10 ===cat("Background (total ranked proteins):", N, "\n")Background (total ranked proteins): 8862 cat("Pathway top 10 genes found in background:", m, "\n")Pathway top 10 genes found in background: 10 cat("Contrast top 10 proteins:", q, "\n")Contrast top 10 proteins: 10 cat("Observed overlap:", k, "\n")Observed overlap: 0 cat("Overlapping genes:", paste(overlap, collapse = ", "), "\n")Overlapping genes: cat("P-value:", p_val, "\n\n")P-value: 1 overlap_df <- data.frame(
Gene = background_genes,
In_Pathway_Top10 = background_genes %in% top10_pathway,
In_Contrast_Top10 = background_genes %in% top10_contrast
) %>%
filter(In_Pathway_Top10 | In_Contrast_Top10) %>%
mutate(Overlap = In_Pathway_Top10 & In_Contrast_Top10) %>%
arrange(desc(Overlap))
print(overlap_df) Gene In_Pathway_Top10 In_Contrast_Top10 Overlap
1 IL34 FALSE TRUE FALSE
2 NNT FALSE TRUE FALSE
3 CX3CL1 FALSE TRUE FALSE
4 MCPT4 FALSE TRUE FALSE
5 RTP1 FALSE TRUE FALSE
6 HYI FALSE TRUE FALSE
7 S100Z FALSE TRUE FALSE
8 TPSAB1 FALSE TRUE FALSE
9 OMP FALSE TRUE FALSE
10 S100A5 FALSE TRUE FALSE
11 SLC6A4 TRUE FALSE FALSE
12 F11R TRUE FALSE FALSE
13 ITGB3 TRUE FALSE FALSE
14 LYN TRUE FALSE FALSE
15 ENTPD1 TRUE FALSE FALSE
16 SYK TRUE FALSE FALSE
17 PLEK TRUE FALSE FALSE
18 PTPN6 TRUE FALSE FALSE
19 FERMT3 TRUE FALSE FALSE
20 P2RY12 TRUE FALSE FALSE