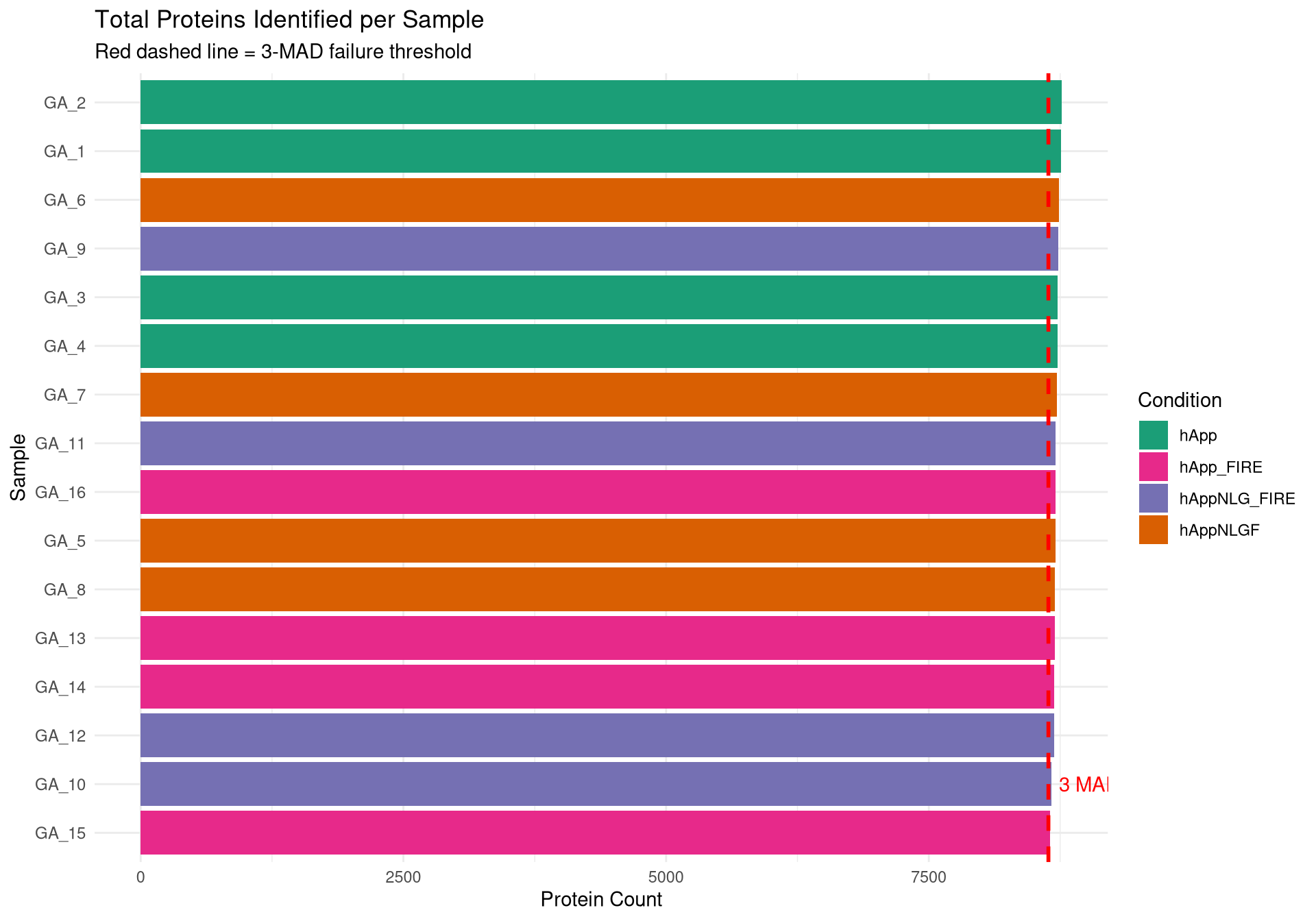
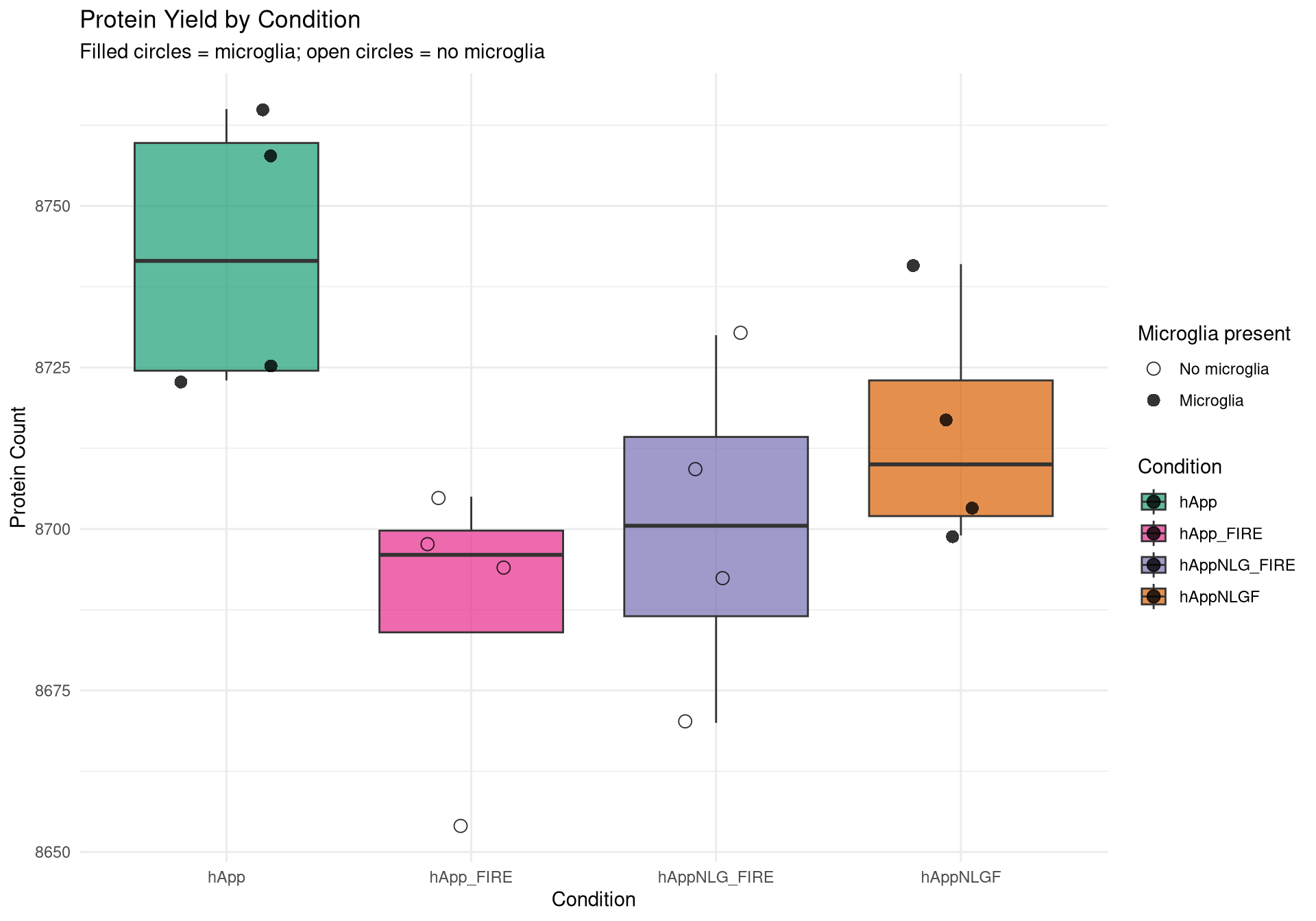
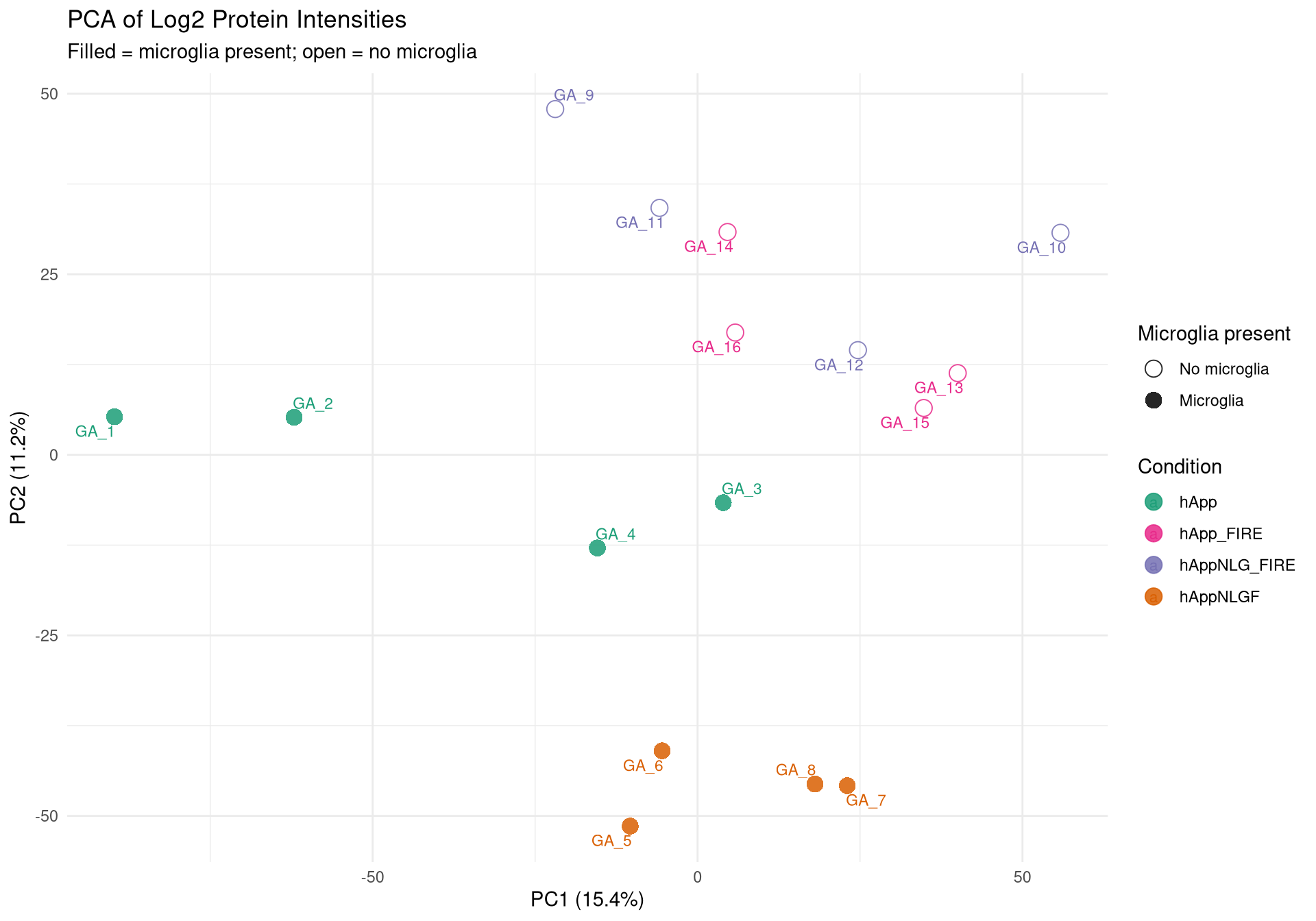
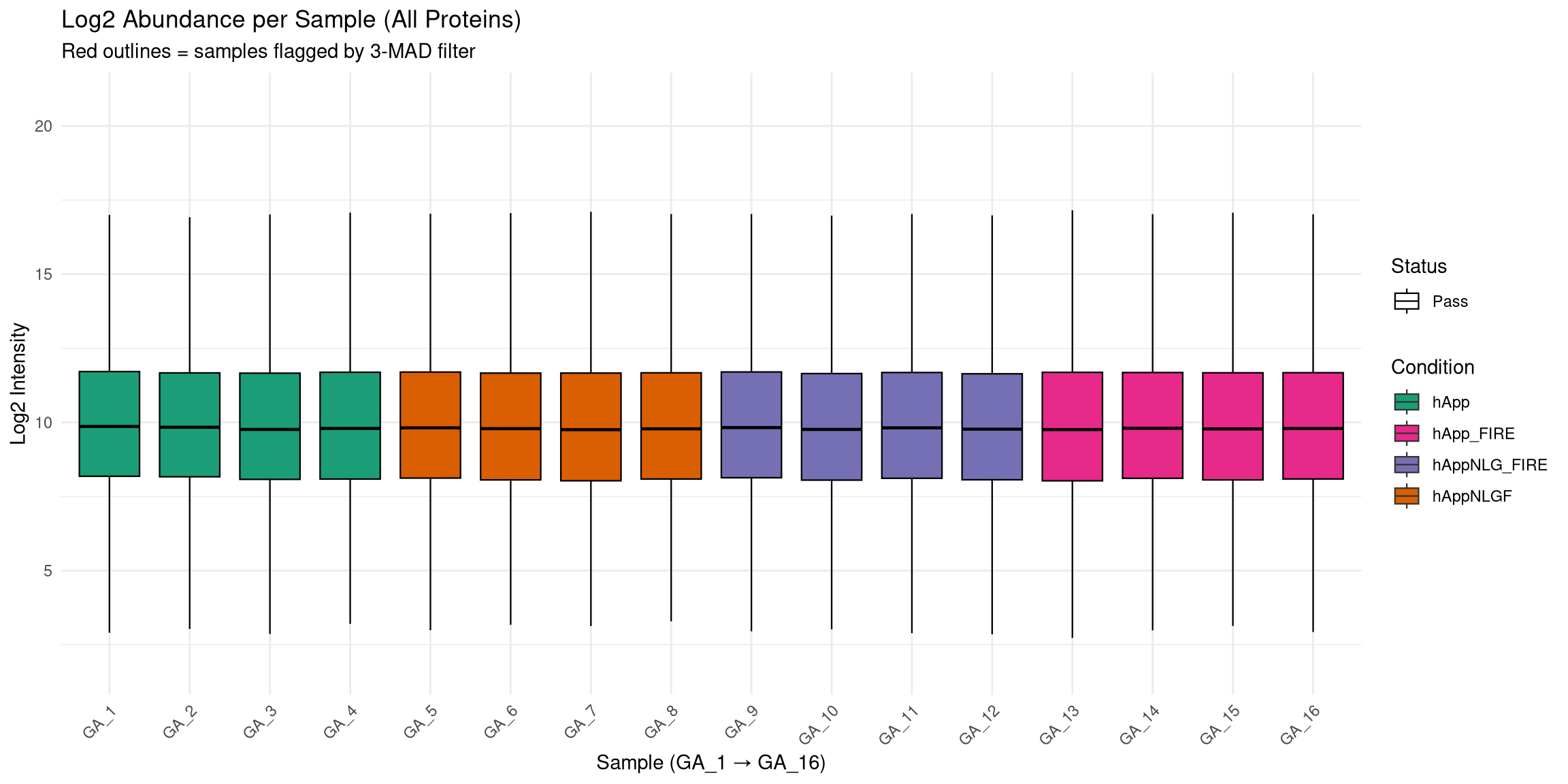
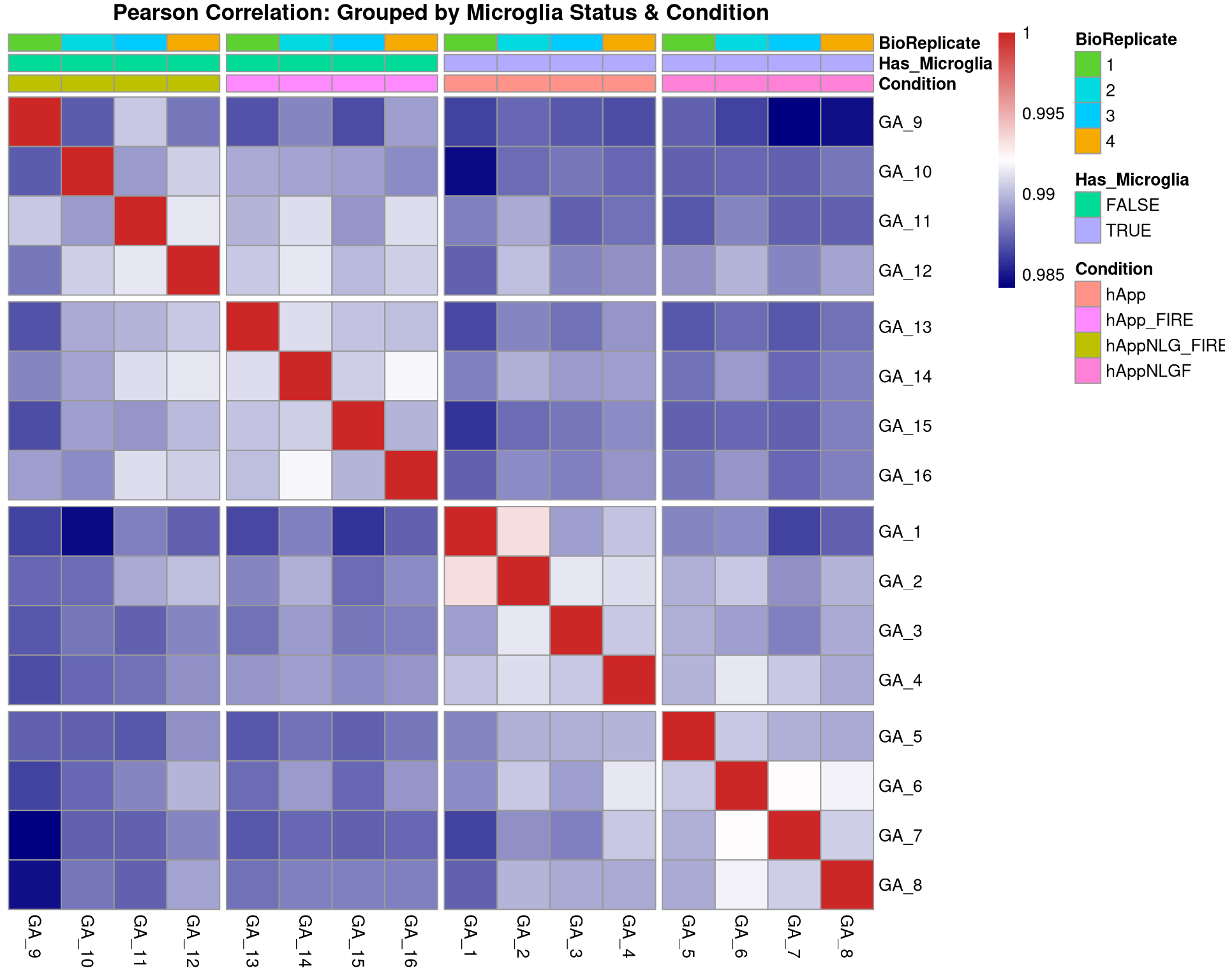
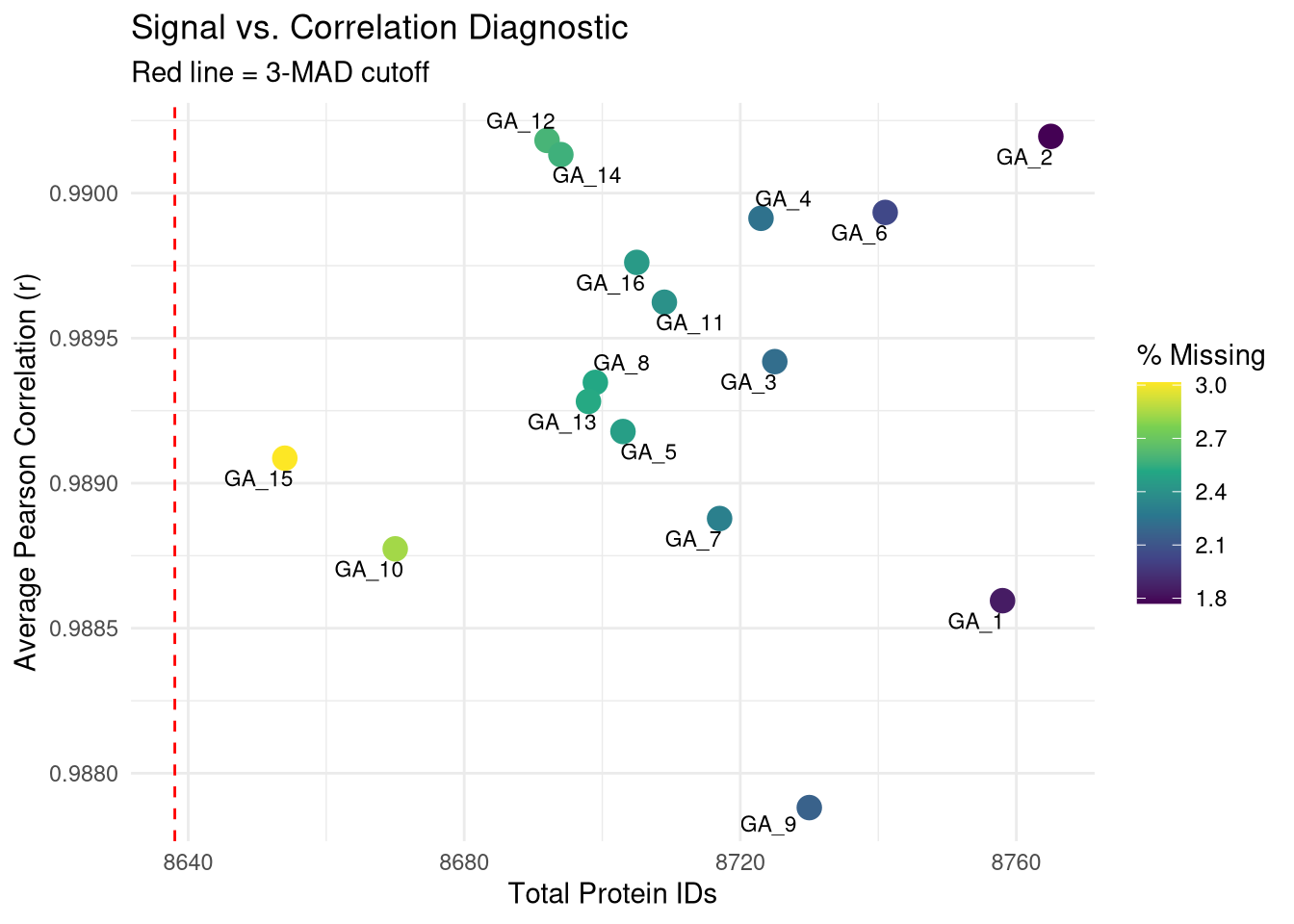
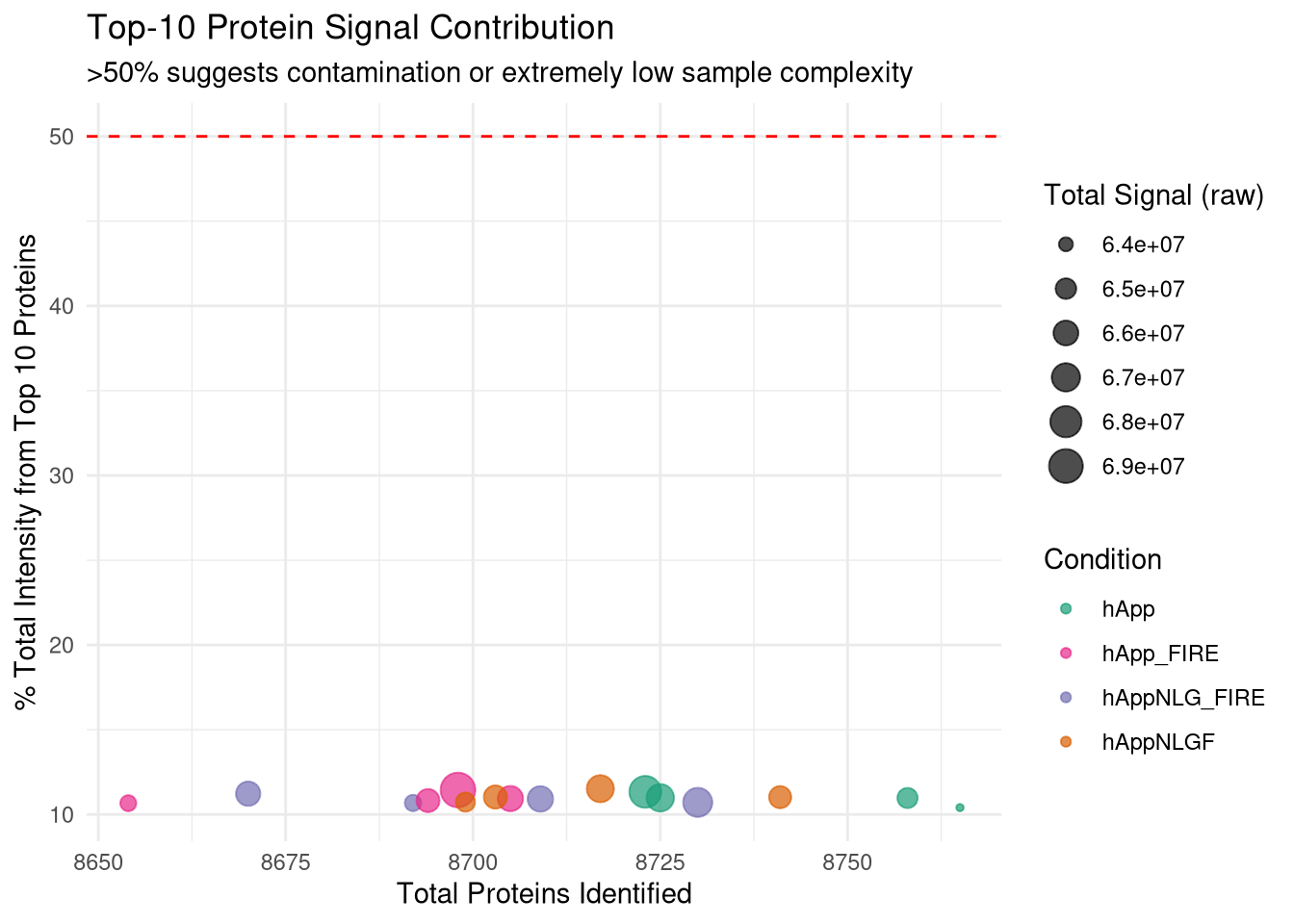
--- title: "Initial QC & Data Cleaning — Proteome" --- ## Libraries ```{r setup, include=FALSE, message=TRUE} library(data.table) library(qs2) library(dplyr) library(ggplot2) library(stringr) library(tidyr) library(pheatmap) ``` ## Set directories ```{r} <- "/nemo/lab/destrooperb/home/shared/zanettc/giulia_proteomics/bulk_proteomics" <- "run4" <- file.path (base_dir, "results" , run_num)<- file.path (base_dir, "data" , "processed" , run_num)<- file.path (base_dir, "input" )dir.create (results_dir, recursive = TRUE , showWarnings = FALSE )dir.create (objects_dir, recursive = TRUE , showWarnings = FALSE )``` ## Define study design ```{r} <- data.table (FileName = paste0 ("GA_" , 1 : 16 ),Condition = rep (c ("hApp" , "hAppNLGF" , "hAppNLG_FIRE" , "hApp_FIRE" ), each = 4 ),BioReplicate = rep (1 : 4 , times = 4 ),Has_Microglia = rep (c (TRUE , TRUE , FALSE , FALSE ), each = 4 )``` ## Read Spectronaut MSStats proteome report ```{r} cat ("Reading Spectronaut report (selecting protein-level columns only)... \n " )#no PG.Genes <- fread (file.path (input_dir, "20260423_154110_PCF000612_GA_MSStats_Report-Proteome.csv" )#, #select = c("R.FileName", "PG.ProteinGroups", "PG.ProteinAccessions", #"PG.Qvalue", "PG.Quantity") cat ("Fragment rows loaded:" , nrow (spec_raw), " \n " )cat ("Unique samples found:" , uniqueN (spec_raw$ R.FileName), " \n " )cat ("Samples present:" , sort (unique (spec_raw$ R.FileName)), " \n " )``` ## Extract unique protein-level quantities per sample ```{r} <- unique (spec_raw[, .(R.FileName, PG.ProteinGroups, PG.ProteinAccessions,cat ("Unique protein-sample rows:" , nrow (pg_long), " \n " )cat ("Unique proteins:" , uniqueN (pg_long$ PG.ProteinGroups), " \n " )``` ## Pivot to wide format (proteins × samples) ```{r} <- dcast (~ R.FileName,value.var = "PG.Quantity" <- paste0 ("GA_" , 1 : 16 )# Sanity-check that all expected samples are present <- setdiff (sample_cols, colnames (quant_wide))if (length (missing_samples) > 0 ) {warning ("These samples are missing from the data: " , paste (missing_samples, collapse = ", " ))else {cat ("All 16 samples found in data. \n " )# Extract quantitative matrix in numeric order (GA_1 … GA_16) <- quant_wide[, ..sample_cols]# Convert Spectronaut NaN to R NA throughout <- as.matrix (quant_data)is.nan (quant_matrix)] <- NA cat ("Proteins in matrix:" , nrow (quant_matrix), " \n " )cat ("Samples in matrix: " , ncol (quant_matrix), " \n " )``` ## Calculate protein ID counts per sample ```{r} <- colSums (! is.na (quant_matrix) & quant_matrix > 0 )<- data.table (FileName = names (protein_counts), Protein_Count = protein_counts)<- merge (qc_counts, meta, by = "FileName" , all.x = TRUE )summary (qc_data$ Protein_Count)``` ## MAD outlier detection ```{r} <- median (qc_data$ Protein_Count, na.rm = TRUE )<- mad (qc_data$ Protein_Count, na.rm = TRUE )<- med_count - (3 * mad_count)cat ("Median Protein Count:" , round (med_count), " \n " )cat ("MAD: " , round (mad_count), " \n " )cat ("3-MAD cutoff: " , round (mad_cutoff), " \n " )<- qc_data[Protein_Count < mad_cutoff, FileName]if (length (failed_runs) > 0 ) {cat (" \n WARNING:" , length (failed_runs), "failed run(s) to drop: \n " )print (failed_runs)else {cat (" \n No runs fell below the 3-MAD threshold. \n " )``` ## Generate PDF QC report ```{r, fig.width=10, fig.height=7} condition_colours <- c( "hApp" = "#1b9e77", "hAppNLGF" = "#d95f02", "hAppNLG_FIRE" = "#7570b3", "hApp_FIRE" = "#e7298a" ) # ── Plot 1: Protein Count per Sample with MAD cutoff ─────────────────────── p1 <- ggplot(qc_data, aes(x = reorder(FileName, Protein_Count), y = Protein_Count, fill = Condition)) + geom_bar(stat = "identity") + geom_hline(yintercept = mad_cutoff, color = "red", linetype = "dashed", linewidth = 1) + annotate("text", x = 2, y = mad_cutoff + 100, label = "3 MAD Cutoff", color = "red", hjust = 0) + scale_fill_manual(values = condition_colours) + coord_flip() + theme_minimal() + labs(title = "Total Proteins Identified per Sample", subtitle = "Red dashed line = 3-MAD failure threshold", x = "Sample", y = "Protein Count") + theme(axis.text.y = element_text(size = 9)) # ── Plot 2: Protein Yield by Condition ───────────────────────────────────── p2 <- ggplot(qc_data, aes(x = Condition, y = Protein_Count, fill = Condition)) + geom_boxplot(outlier.shape = NA, alpha = 0.7) + geom_point(aes(shape = Has_Microglia), position = position_jitter(width = 0.2), alpha = 0.8, size = 3) + scale_fill_manual(values = condition_colours) + scale_shape_manual(values = c("TRUE" = 16, "FALSE" = 1), labels = c("TRUE" = "Microglia", "FALSE" = "No microglia")) + theme_minimal() + labs(title = "Protein Yield by Condition", subtitle = "Filled circles = microglia; open circles = no microglia", x = "Condition", y = "Protein Count", shape = "Microglia present") # ── PCA ───────────────────────────────────────────────────────────────────── cat("Preparing PCA...\n") pca_mat_log <- log2(quant_matrix + 1) pca_mat_log_t <- t(pca_mat_log) noise_floor <- min(pca_mat_log_t[pca_mat_log_t > 0], na.rm = TRUE) / 2 pca_mat_log_t[is.na(pca_mat_log_t) | pca_mat_log_t == 0] <- noise_floor valid_cols <- apply(pca_mat_log_t, 2, var) > 0 pca_mat_log_t <- pca_mat_log_t[, valid_cols] cat("Proteins retained for PCA (non-zero variance):", sum(valid_cols), "\n") pca_res <- prcomp(pca_mat_log_t, center = TRUE, scale. = TRUE) var_expl <- round(100 * pca_res$sdev^2 / sum(pca_res$sdev^2), 1) pca_df <- as.data.frame(pca_res$x) pca_df$FileName <- rownames(pca_df) pca_df <- merge(pca_df, meta, by = "FileName", all.x = TRUE) # ── Plot 3: PCA by Condition ──────────────────────────────────────────────── p3 <- ggplot(pca_df, aes(x = PC1, y = PC2, color = Condition, shape = Has_Microglia)) + geom_point(size = 4, alpha = 0.85) + ggrepel::geom_text_repel(aes(label = FileName), size = 3, max.overlaps = 20) + scale_color_manual(values = condition_colours) + scale_shape_manual(values = c("TRUE" = 16, "FALSE" = 1), labels = c("TRUE" = "Microglia", "FALSE" = "No microglia")) + theme_minimal() + labs(title = "PCA of Log2 Protein Intensities", subtitle = "Filled = microglia present; open = no microglia", x = paste0("PC1 (", var_expl[1], "%)"), y = paste0("PC2 (", var_expl[2], "%)"), shape = "Microglia present") # ── Save PDF ──────────────────────────────────────────────────────────────── pdf_path <- file.path(results_dir, "Initial_QC_Report.pdf") cat("Saving PDF:", pdf_path, "\n") pdf(pdf_path, width = 10, height = 7) print(p1); print(p2); print(p3) dev.off() print(p1); print(p2); print(p3) ``` ## Abundance distributions ```{r, fig.width=12, fig.height=6} quant_log_plot <- log2(quant_matrix + 1) quant_log_plot[quant_log_plot == 0] <- NA abund_long <- as.data.frame(quant_log_plot) %>% mutate(Protein = row_number()) %>% pivot_longer(-Protein, names_to = "FileName", values_to = "Log2_Intensity") %>% drop_na() %>% left_join(as.data.frame(qc_data), by = "FileName") %>% mutate( Status = ifelse(FileName %in% failed_runs, "Failed (Outlier)", "Pass"), Sample_Num = as.integer(str_extract(FileName, "[0-9]+")) ) p_abund <- ggplot(abund_long, aes(x = reorder(FileName, Sample_Num), y = Log2_Intensity, fill = Condition)) + geom_boxplot(aes(color = Status), outlier.shape = NA, lwd = 0.4) + scale_fill_manual(values = condition_colours) + scale_color_manual(values = c("Pass" = "black", "Failed (Outlier)" = "red")) + theme_minimal() + labs(title = "Log2 Abundance per Sample (All Proteins)", subtitle = "Red outlines = samples flagged by 3-MAD filter", x = "Sample (GA_1 → GA_16)", y = "Log2 Intensity") + theme(axis.text.x = element_text(angle = 45, hjust = 1)) print(p_abund) ``` ## Correlation heatmap ```{r, fig.width=10, fig.height=8} quant_mat_cor <- log2(quant_matrix + 1) quant_mat_cor[quant_mat_cor == 0] <- NA cor_mat <- cor(quant_mat_cor, use = "pairwise.complete.obs") meta_sorted <- meta %>% arrange(Has_Microglia, Condition, BioReplicate) %>% as.data.frame() sorted_runs <- meta_sorted$FileName cor_mat_sorted <- cor_mat[sorted_runs, sorted_runs] anno_df <- meta_sorted %>% select(FileName, Condition, Has_Microglia, BioReplicate) %>% mutate(Has_Microglia = as.factor(Has_Microglia), BioReplicate = as.factor(BioReplicate)) %>% as.data.frame() rownames(anno_df) <- anno_df$FileName anno_df$FileName <- NULL pheatmap(cor_mat_sorted, main = "Pearson Correlation: Grouped by Microglia Status & Condition", annotation_col = anno_df, cluster_rows = FALSE, cluster_cols = FALSE, show_rownames = TRUE, show_colnames = TRUE, color = colorRampPalette(c("navy", "white", "firebrick3"))(100), gaps_col = which(diff(as.numeric(as.factor(meta_sorted$Condition))) != 0), gaps_row = which(diff(as.numeric(as.factor(meta_sorted$Condition))) != 0)) ``` ## Average-correlation diagnostic ```{r} <- data.table (FileName = colnames (quant_mat_cor),Protein_Count = protein_counts[colnames (quant_mat_cor)],NA_Percent = colMeans (is.na (quant_mat_cor)) * 100 ,Avg_Correlation = rowMeans (cor_mat, na.rm = TRUE )<- qc_diagnostics[which.min (Avg_Correlation), FileName]cat ("--- Diagnostic for lowest-correlation sample --- \n " )print (qc_diagnostics[FileName == outlier_id])<- ggplot (qc_diagnostics, aes (x = Protein_Count, y = Avg_Correlation)) + geom_point (aes (color = NA_Percent), size = 4 ) + geom_vline (xintercept = mad_cutoff, linetype = "dashed" , color = "red" ) + :: geom_text_repel (aes (label = FileName), size = 3 , max.overlaps = 20 ) + scale_color_viridis_c (name = "% Missing" ) + theme_minimal () + labs (title = "Signal vs. Correlation Diagnostic" ,subtitle = "Red line = 3-MAD cutoff" ,x = "Total Protein IDs" , y = "Average Pearson Correlation (r)" )print (p_diag)``` ## Contaminant check: top-10 protein signal contribution ```{r} <- function (column, n = 10 ) {<- sort (column, decreasing = TRUE , na.last = TRUE )sum (sorted_vals[1 : n], na.rm = TRUE ) / sum (column, na.rm = TRUE )) * 100 <- apply (quant_matrix, 2 , calc_top_n_pct, n = 10 ): = top10_metrics[FileName]]: = colSums (quant_matrix, na.rm = TRUE )[FileName]]<- qc_data[Top10_Percent > 50 ,cat ("Samples where top 10 proteins account for >50% of signal: \n " )print (contaminant_suspects)<- ggplot (qc_data, aes (x = Protein_Count, y = Top10_Percent)) + geom_point (aes (color = Condition, size = Total_Intensity), alpha = 0.7 ) + geom_hline (yintercept = 50 , linetype = "dashed" , color = "red" ) + :: geom_text_repel (aes (label = ifelse (Top10_Percent > 50 , FileName, "" )),size = 3 , max.overlaps = 10 + scale_color_manual (values = condition_colours) + theme_minimal () + labs (title = "Top-10 Protein Signal Contribution" ,subtitle = ">50% suggests contamination or extremely low sample complexity" ,x = "Total Proteins Identified" ,y = "% Total Intensity from Top 10 Proteins" ,size = "Total Signal (raw)" )print (p_top10)``` ## Identify top contributing proteins in suspect samples ```{r} if (nrow (contaminant_suspects) > 0 ) {<- contaminant_suspects$ FileName<- apply (quant_matrix[, suspect_cols, drop = FALSE ], 2 , function (x) {$ PG.ProteinGroups[which.max (x)]cat ("Top protein per suspect sample: \n " )print (top_ids)else {cat ("No suspect samples to investigate. \n " )``` ## Filter out failed samples ```{r} <- meta$ FileName<- setdiff (valid_runs, failed_runs)cat ("Original samples: " , length (valid_runs), " \n " )cat ("Samples dropped (3-MAD): " , length (failed_runs), " \n " )cat ("Clean samples remaining: " , length (final_runs), " \n " )if (length (failed_runs) > 0 ) print (failed_runs)``` ## Save objects ```{r} # Clean wide protein matrix (proteins × passing samples) <- cbind (# Clean metadata <- qc_data[FileName %in% final_runs]# Dropped sample log <- qc_data[FileName %in% failed_runs,: = "Sub 3-MAD Protein Count" ]fwrite (spec_raw, file.path (objects_dir, "clean_spec_raw.csv" ))fwrite (clean_meta, file.path (objects_dir, "clean_metadata.csv" ))fwrite (clean_quant_wide, file.path (objects_dir, "clean_quant_wide.csv" ))fwrite (dropped_log, file.path (objects_dir, "dropped_samples_log.csv" ))cat ("Objects saved to:" , objects_dir, " \n " )```