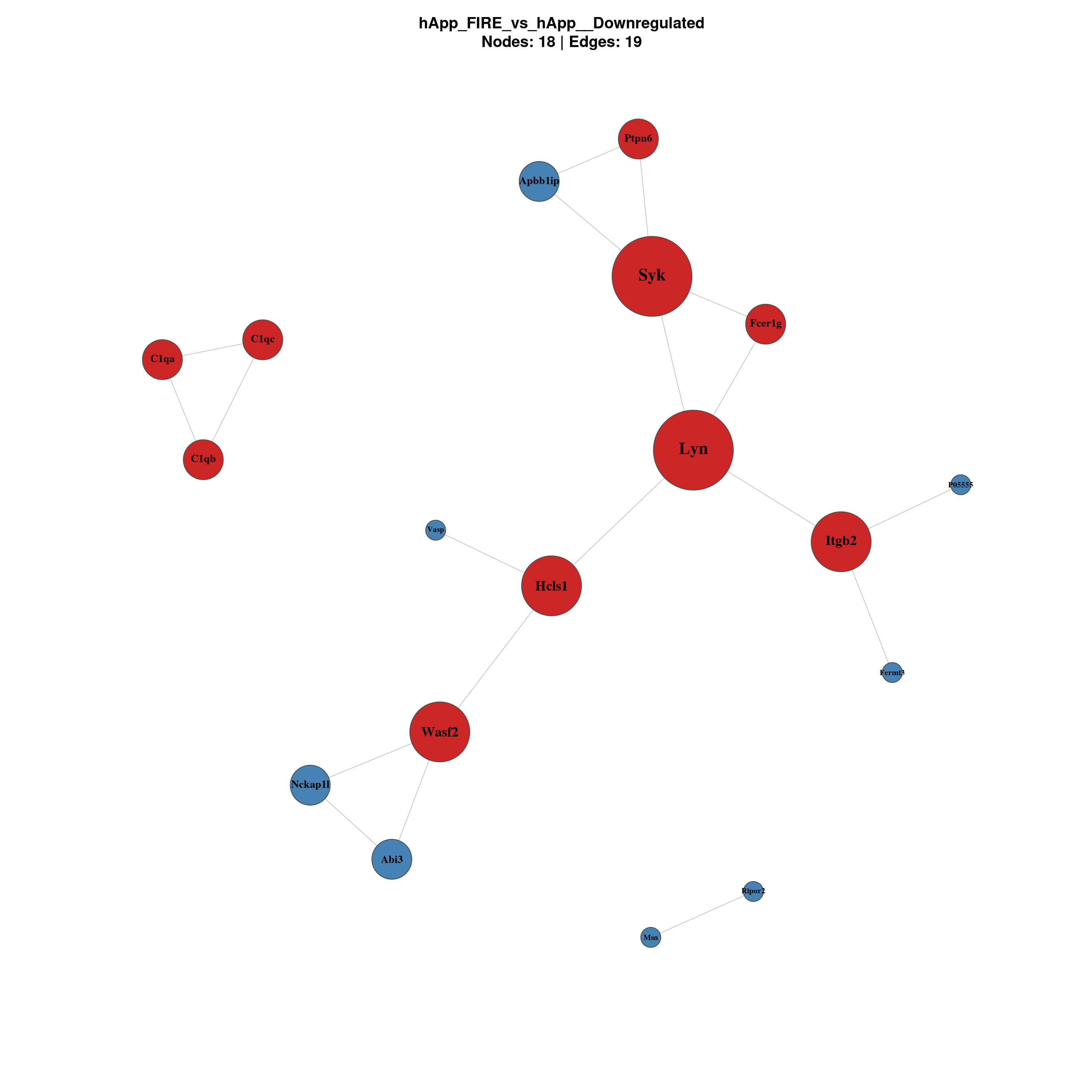
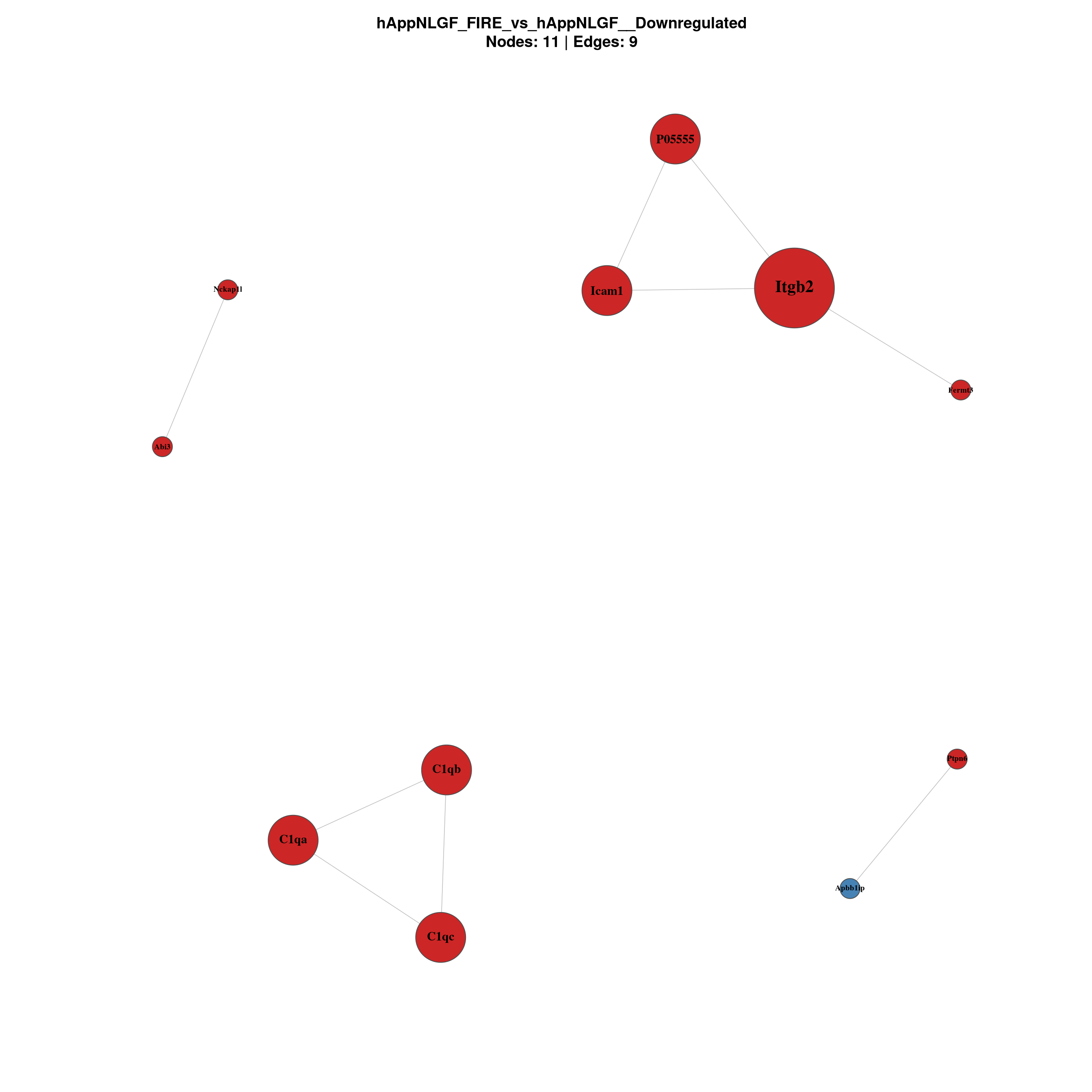
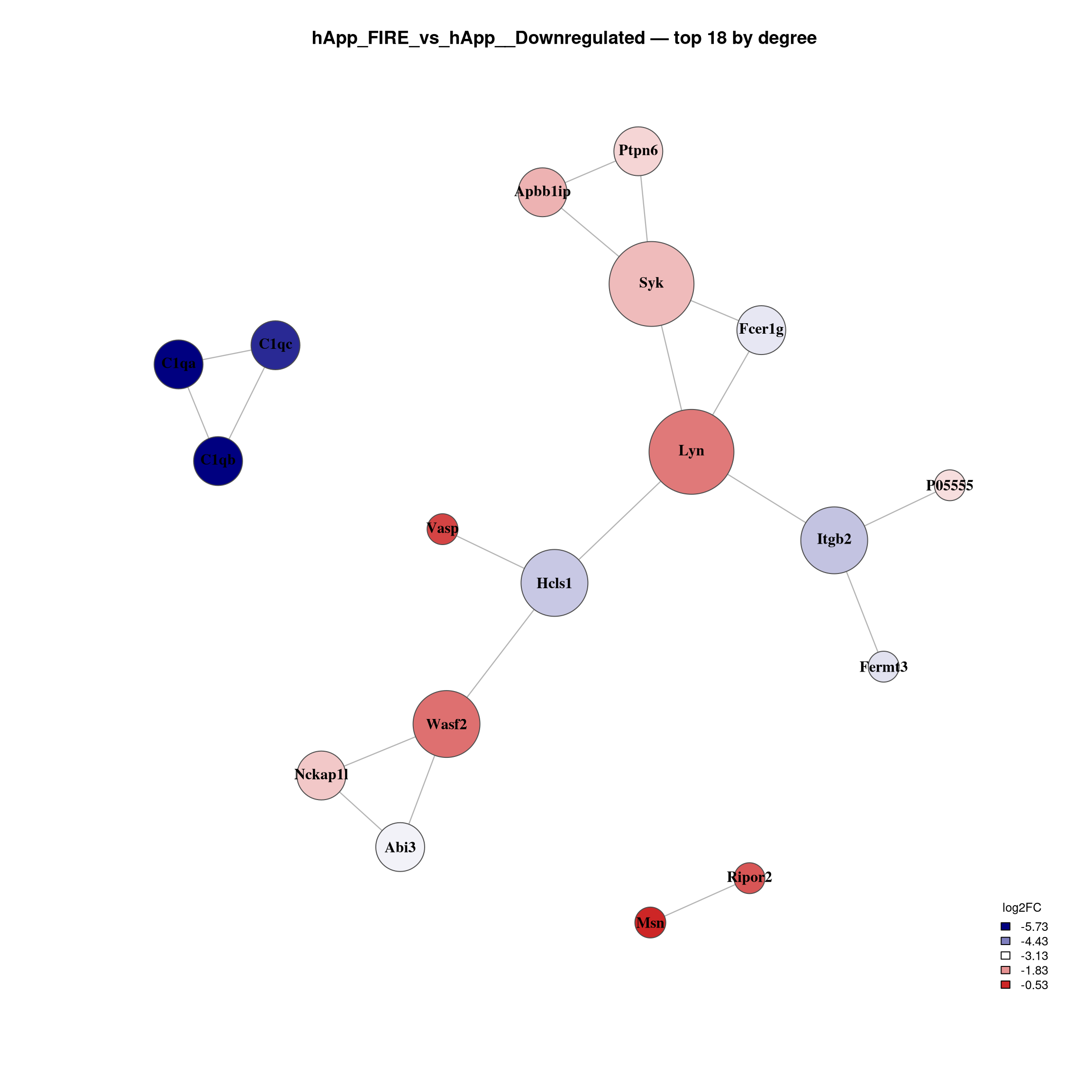
--- title: "6. PPI Network Analysis" format: html --- ## Overview `millie_proteomics/notebooks/08_PPI.qmd` , with this project's contrast set (four pairwise + the APP × Microglia interaction).## Libraries ```{r setup, message=TRUE, warning=FALSE,} library(data.table) library(dplyr) library(tidyr) library(ggplot2) library(ggrepel) library(stringr) library(STRINGdb) library(igraph) library(qs2) library(scales) ``` ## Directories and parameters ```{r} <- "/nemo/lab/destrooperb/home/shared/zanettc/giulia_proteomics/bulk_proteomics" <- "run4" <- file.path (base_dir, "results" , run_num)<- file.path (base_dir, "data" , "processed" , run_num)<- file.path (results_dir, "PPI_networks" )dir.create (ppi_dir, recursive = TRUE , showWarnings = FALSE )<- 0.05 <- 0.5 <- 700 <- 30 # nodes shown in the top-N subnetwork plot ``` ## Load and prepare DE results ```{r} <- fread (file.path (results_dir, "Full_GroupComparison_Results.csv" ))<- fread (file.path (objects_dir, "protein_dictionary.csv" ))<- res_df %>% filter (! is.na (adj.pvalue), is.finite (log2FC)) %>% left_join (protein_dictionary, by = "Protein" ) %>% mutate (Gene = ifelse (is.na (Gene) | Gene == "" , Protein, Gene),Status = case_when (< FDR_CUTOFF & log2FC > LOGFC_CUTOFF ~ "Upregulated" ,< FDR_CUTOFF & log2FC < - LOGFC_CUTOFF ~ "Downregulated" ,TRUE ~ "Not Significant" ``` ## Initialise STRINGdb (mouse, physical, high-confidence) ```{r} # Crick cluster blocks HTTPS with SSL; wget --no-check-certificate bypasses it options (download.file.method = "wget" ,download.file.extra = "--no-check-certificate" )<- "/nemo/lab/destrooperb/home/shared/zanettc/giulia_proteomics/bulk_proteomics/data/stringdb_cache" dir.create (stringdb_cache, recursive = TRUE , showWarnings = FALSE )<- STRINGdb$ new (version = "12.0" ,species = 10090 ,score_threshold = STRING_SCORE,network_type = "physical" ,input_directory = stringdb_cache``` ## Build networks per contrast and direction ```{r} <- c ("hAppNLGF_vs_hApp" ,"hApp_FIRE_vs_hApp" ,"hAppNLGF_FIRE_vs_hAppNLGF" ,"hAppNLGF_FIRE_vs_hApp_FIRE" ,"Interaction_APP_x_Microglia" <- function (genes_df) {if (nrow (genes_df) < 5 ) return (NULL )<- string_db$ map (genes_df, "Gene" , removeUnmappedRows = TRUE )if (nrow (mapped) < 5 ) return (NULL )<- string_db$ get_interactions (mapped$ STRING_id)if (nrow (inter) == 0 )return (list (proteins = mapped, interactions = inter, graph = NULL ))<- graph_from_data_frame (%>% dplyr:: select (from, to, combined_score),directed = FALSE ,vertices = mapped %>% dplyr:: select (STRING_id, Gene, log2FC)%>% simplify ()V (g)$ degree <- degree (g)list (proteins = mapped, interactions = inter, graph = g)<- list ()for (comp in contrasts) {cat (" \n ============================ \n " , comp, " \n ============================ \n " )for (dir_lab in c ("Upregulated" , "Downregulated" )) {<- msstats_ready %>% filter (Label == comp, Status == dir_lab) %>% :: select (Gene, log2FC, adj.pvalue) %>% distinct (Gene, .keep_all = TRUE ) %>% as.data.frame ()cat (sprintf (" %-13s n = %d \n " , dir_lab, nrow (sig)))<- build_network (sig)if (! is.null (nr) && ! is.null (nr$ graph))cat (sprintf (" -> nodes %d | edges %d \n " ,vcount (nr$ graph), ecount (nr$ graph)))paste (comp, dir_lab, sep = "__" )]] <- nr``` ## Visualise full networks (one PDF per contrast/direction) ```{r plot_full, fig.width=14, fig.height=14} plot_full_network <- function(nr, label) { if (is.null(nr) || is.null(nr$graph) || ecount(nr$graph) == 0) return(invisible()) g_plot <- delete_vertices(nr$graph, V(nr$graph)[degree(nr$graph) == 0]) if (vcount(g_plot) < 3) return(invisible()) V(g_plot)$label <- V(g_plot)$Gene V(g_plot)$size <- rescale(degree(g_plot), to = c(5, 20)) V(g_plot)$label.cex <- rescale(degree(g_plot), to = c(0.6, 1.3)) V(g_plot)$label.font <- 2 V(g_plot)$color <- "steelblue" V(g_plot)$frame.color <- "grey30" E(g_plot)$color <- "grey75" E(g_plot)$width <- 0.8 top_hubs <- names(sort(degree(g_plot), decreasing = TRUE))[1:min(10, vcount(g_plot))] V(g_plot)$color[V(g_plot)$name %in% top_hubs] <- "firebrick3" pdf(file.path(ppi_dir, paste0("network_full_", label, ".pdf")), width = 14, height = 14) set.seed(42) plot(g_plot, layout = layout_with_fr(g_plot), vertex.label.color = "black", main = paste0(label, "\nSTRING physical >= ", STRING_SCORE, " | Nodes: ", vcount(g_plot), " | Edges: ", ecount(g_plot), " | Red = top 10 hubs")) dev.off() set.seed(42) plot(g_plot, layout = layout_with_fr(g_plot), vertex.label.color = "black", main = paste0(label, "\nNodes: ", vcount(g_plot), " | Edges: ", ecount(g_plot))) } for (key in names(network_results)) plot_full_network(network_results[[key]], key) ``` ## Top-N subnetwork plots — focused view of strongest hubs ```{r plot_topN, fig.width=12, fig.height=12} plot_top_network <- function(nr, label, top_n = TOP_N) { if (is.null(nr) || is.null(nr$graph) || ecount(nr$graph) == 0) return(invisible()) g <- nr$graph top_nodes <- names(sort(degree(g), decreasing = TRUE))[1:min(top_n, vcount(g))] g_top <- induced_subgraph(g, top_nodes) %>% (function(x) delete_vertices(x, V(x)[degree(x) == 0]))() if (vcount(g_top) < 3) return(invisible()) V(g_top)$label <- V(g_top)$Gene V(g_top)$size <- rescale(degree(g_top), to = c(8, 22)) V(g_top)$label.cex <- 1.0 V(g_top)$label.font <- 2 V(g_top)$frame.color <- "grey30" E(g_top)$color <- "grey70" E(g_top)$width <- 1.2 fc <- V(g_top)$log2FC pal <- colorRampPalette(c("navy", "white", "firebrick3"))(100) fc_idx <- findInterval(fc, seq(min(fc, na.rm = TRUE), max(fc, na.rm = TRUE), length.out = 100)) fc_idx[fc_idx == 0] <- 1 V(g_top)$color <- pal[fc_idx] pdf(file.path(ppi_dir, paste0("network_top", top_n, "_", label, ".pdf")), width = 12, height = 12) set.seed(42) plot(g_top, layout = layout_with_fr(g_top), vertex.label.color = "black", main = paste0(label, " — top ", vcount(g_top), " by degree\n", "Coloured by log2FC | Edges: ", ecount(g_top))) legend_vals <- seq(min(fc, na.rm = TRUE), max(fc, na.rm = TRUE), length.out = 5) legend_colors <- colorRampPalette(c("navy", "white", "firebrick3"))(5) legend("bottomright", legend = round(legend_vals, 2), fill = legend_colors, title = "log2FC", bty = "n", cex = 0.8) dev.off() set.seed(42) plot(g_top, layout = layout_with_fr(g_top), vertex.label.color = "black", main = paste0(label, " — top ", vcount(g_top), " by degree")) legend("bottomright", legend = round(legend_vals, 2), fill = legend_colors, title = "log2FC", bty = "n", cex = 0.8) } for (key in names(network_results)) plot_top_network(network_results[[key]], key) ``` ## Hub protein summary tables ```{r} <- list ()for (key in names (network_results)) {<- network_results[[key]]if (is.null (nr) || is.null (nr$ graph)) next <- nr$ graph<- data.frame (Gene = V (g)$ Gene,log2FC = V (g)$ log2FC,Degree = degree (g),Comparison = key%>% filter (Degree > 0 ) %>% arrange (desc (Degree))<- hub_dffwrite (hub_df, file.path (ppi_dir, paste0 ("hub_proteins_" , key, ".csv" )))cat (" \n --- Top 20 hubs:" , key, "--- \n " )print (head (hub_df, 20 ))if (length (hub_list) > 0 ) {<- bind_rows (hub_list)fwrite (hub_combined, file.path (ppi_dir, "hub_proteins_all.csv" ))``` ## Network summary statistics ```{r} <- data.frame (Comparison = character (),Direction = character (),Sig_proteins = integer (),Mapped_to_STRING = integer (),Nodes_with_edges = integer (),Edges = integer (),stringsAsFactors = FALSE for (key in names (network_results)) {<- strsplit (key, "__" , fixed = TRUE )[[1 ]]<- parts[1 ]; dir_lab <- parts[2 ]<- network_results[[key]]<- if (! is.null (nr)) nr$ graph else NULL <- if (! is.null (g)) sum (degree (g) > 0 ) else 0 <- if (! is.null (g)) ecount (g) else 0 <- nrow (msstats_ready %>% filter (Label == comp, Status == dir_lab) %>% distinct (Gene))<- rbind (summary_table, data.frame (Comparison = comp,Direction = dir_lab,Sig_proteins = sig_n,Mapped_to_STRING = if (! is.null (nr)) nrow (nr$ proteins) else 0 ,Nodes_with_edges = n_with_edges,Edges = n_edgesprint (summary_table)fwrite (summary_table, file.path (ppi_dir, "network_summary.csv" ))``` ## Session info ```{r} sessionInfo ()```