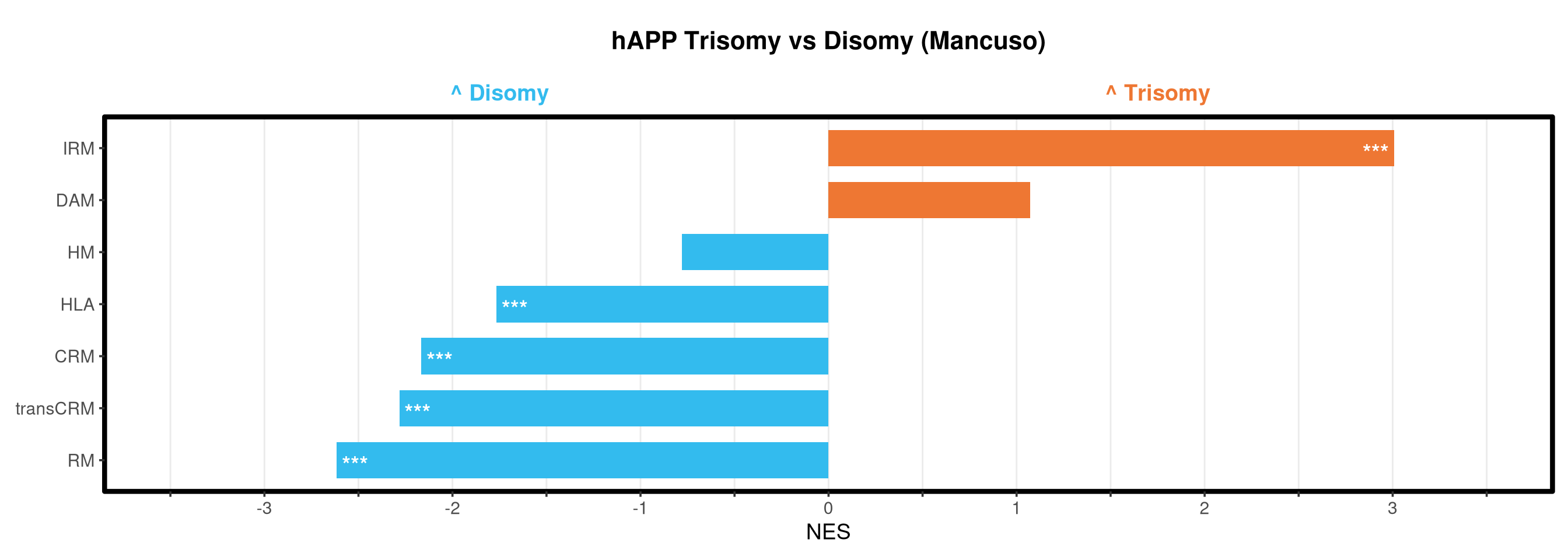
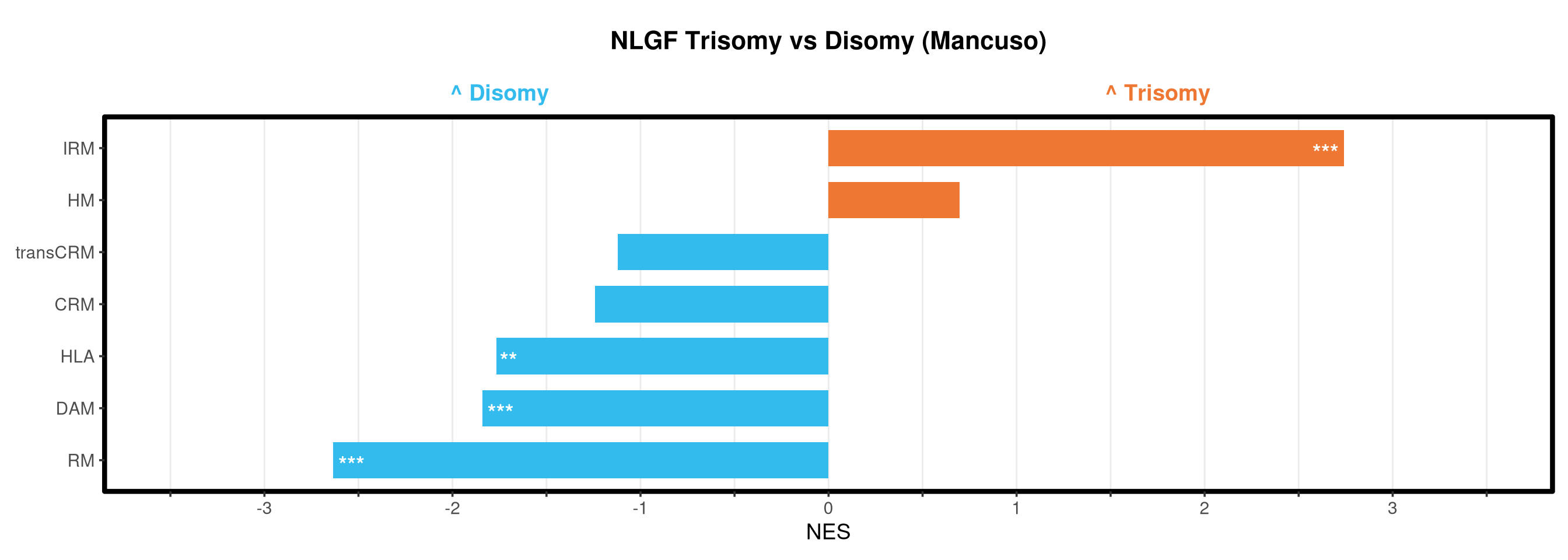
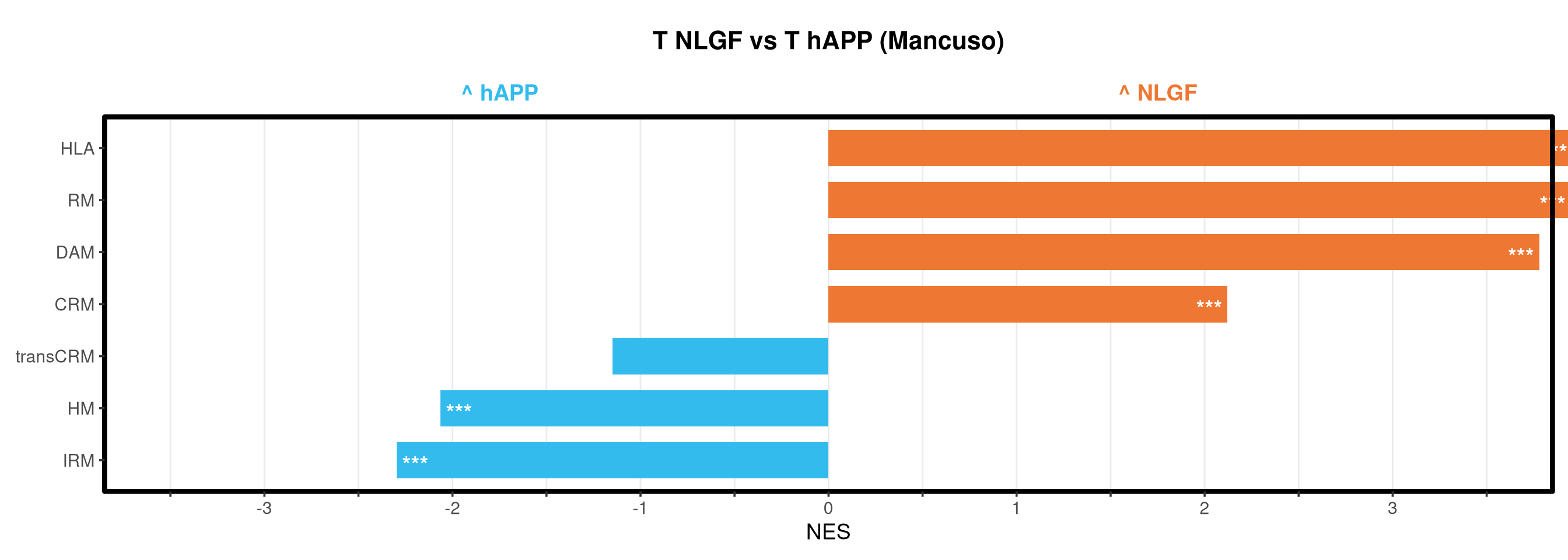
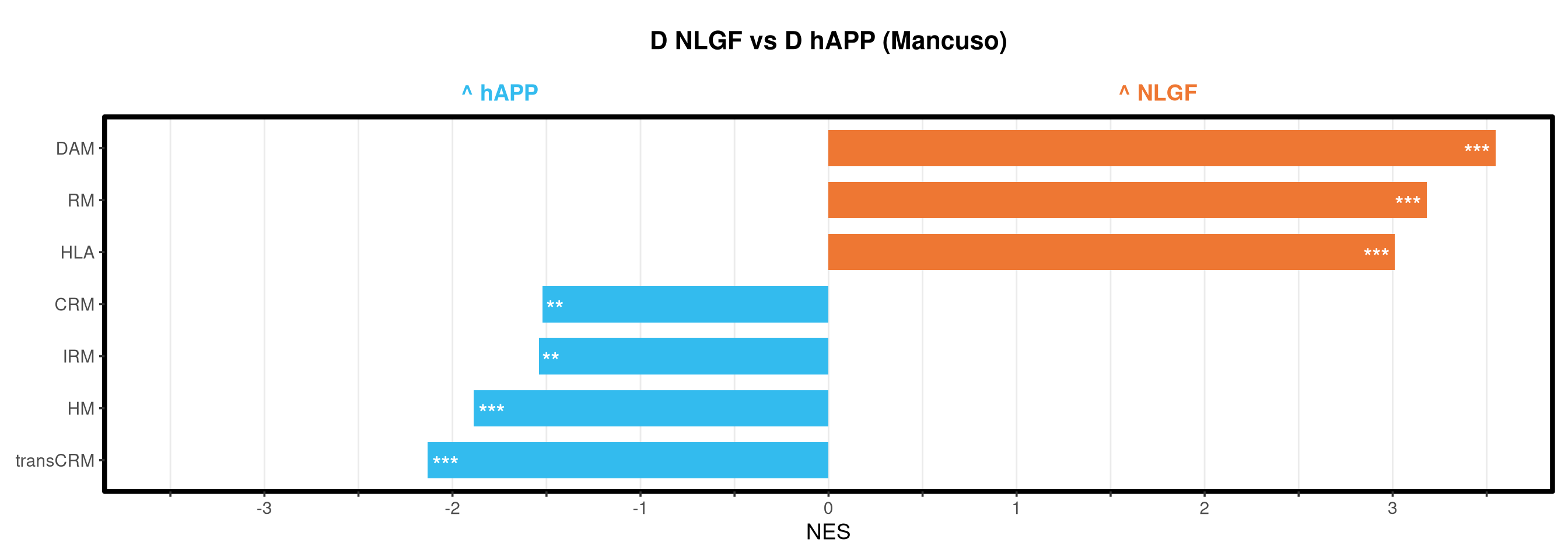
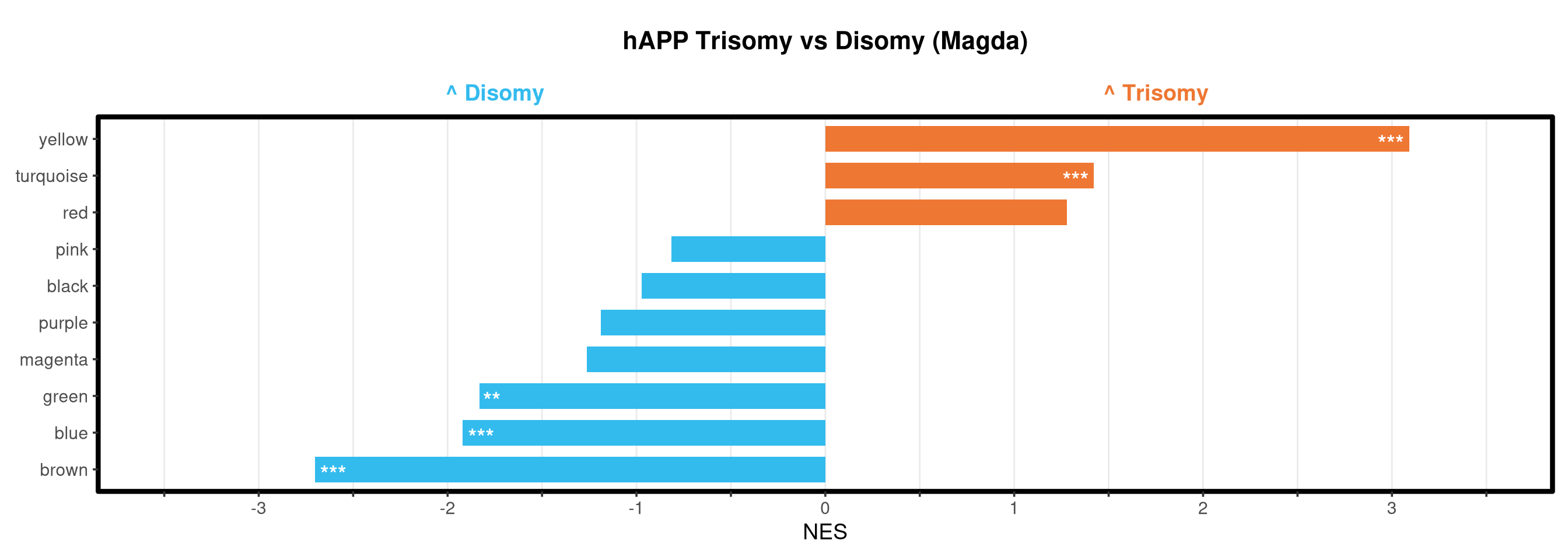
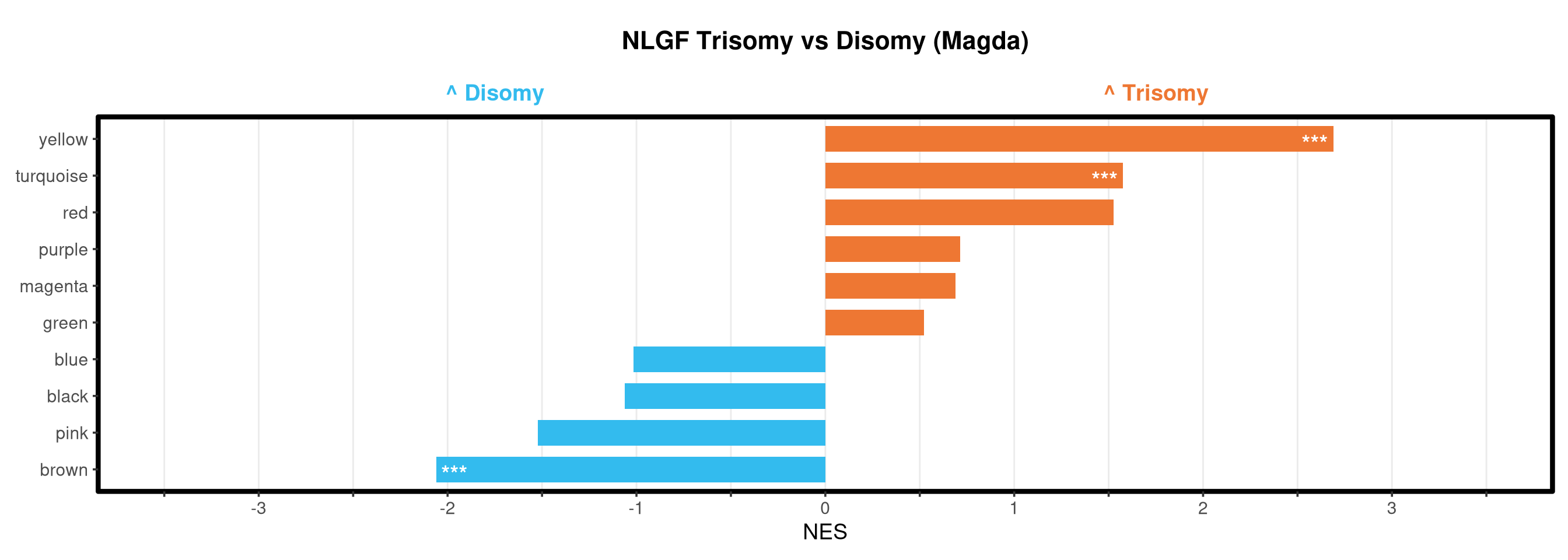
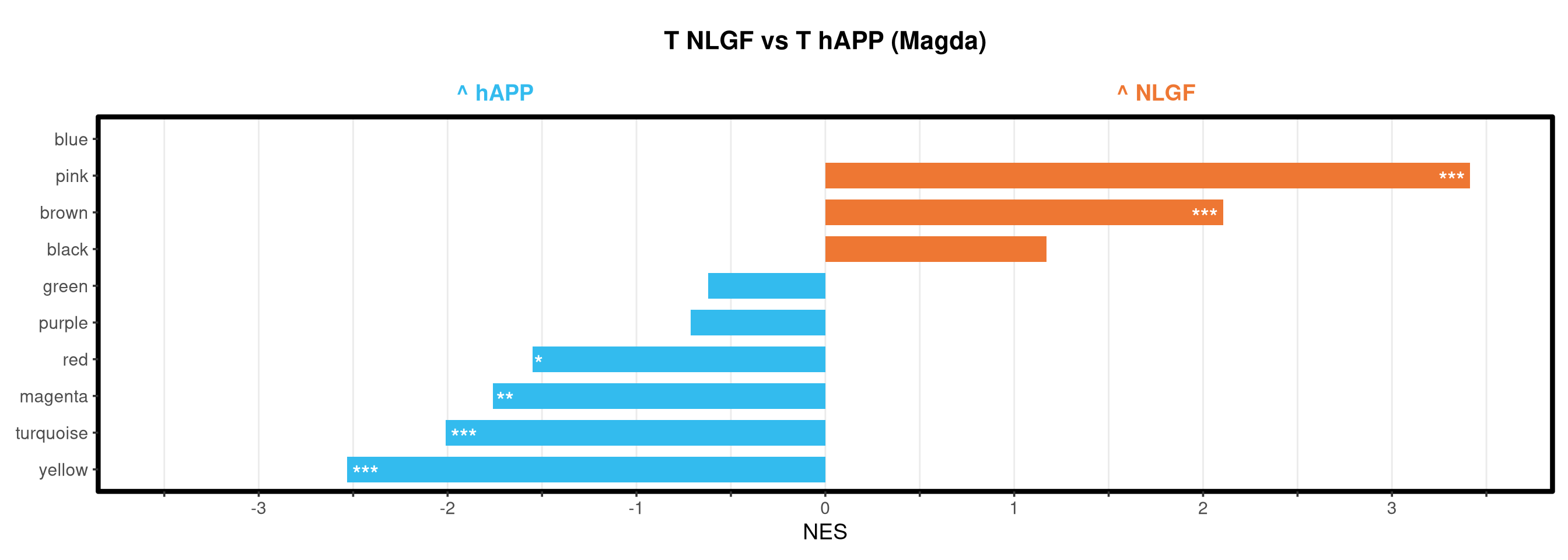
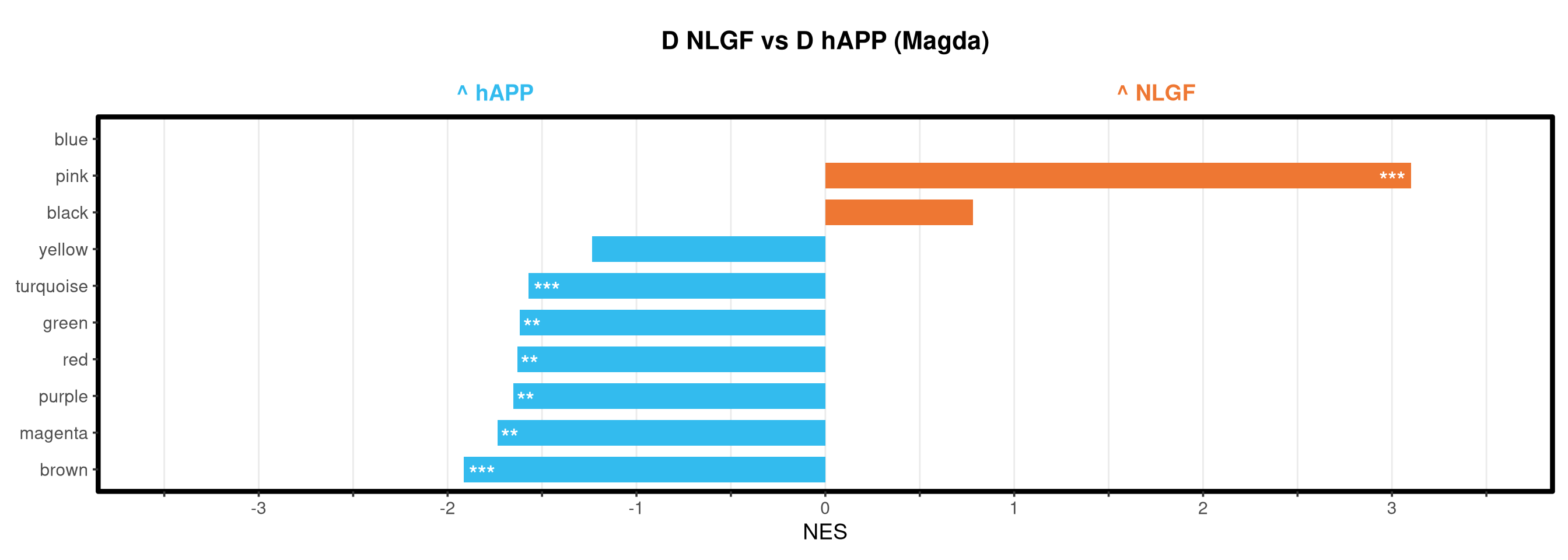
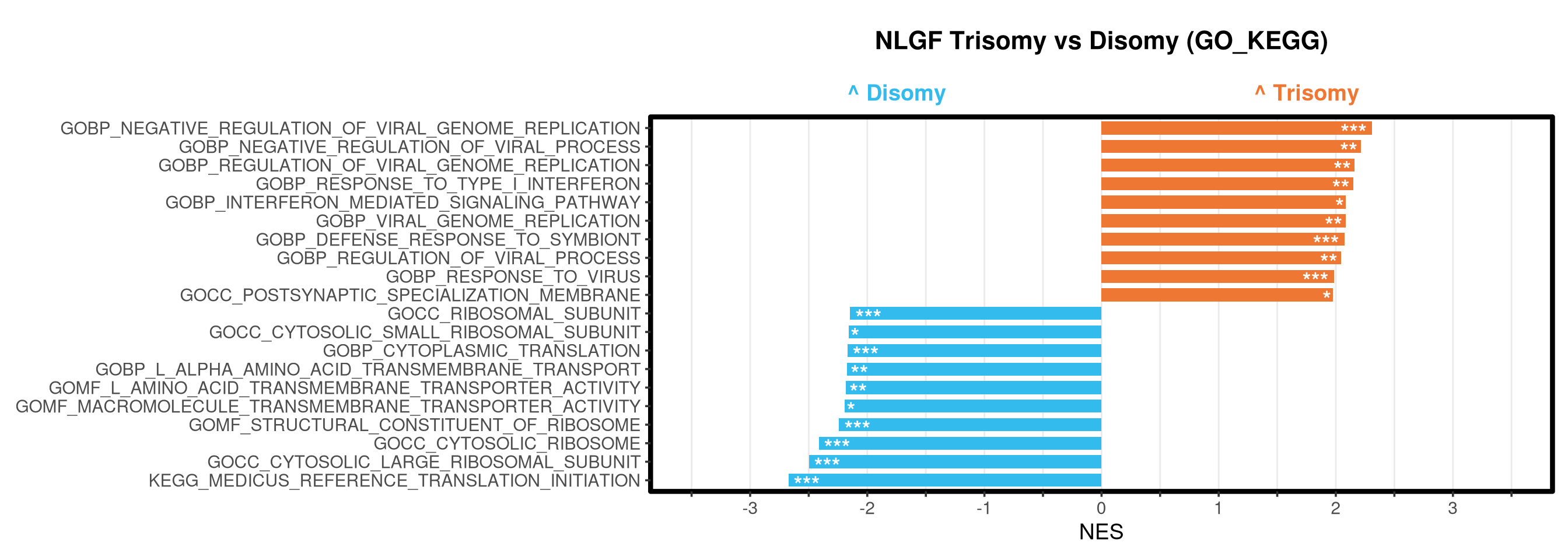
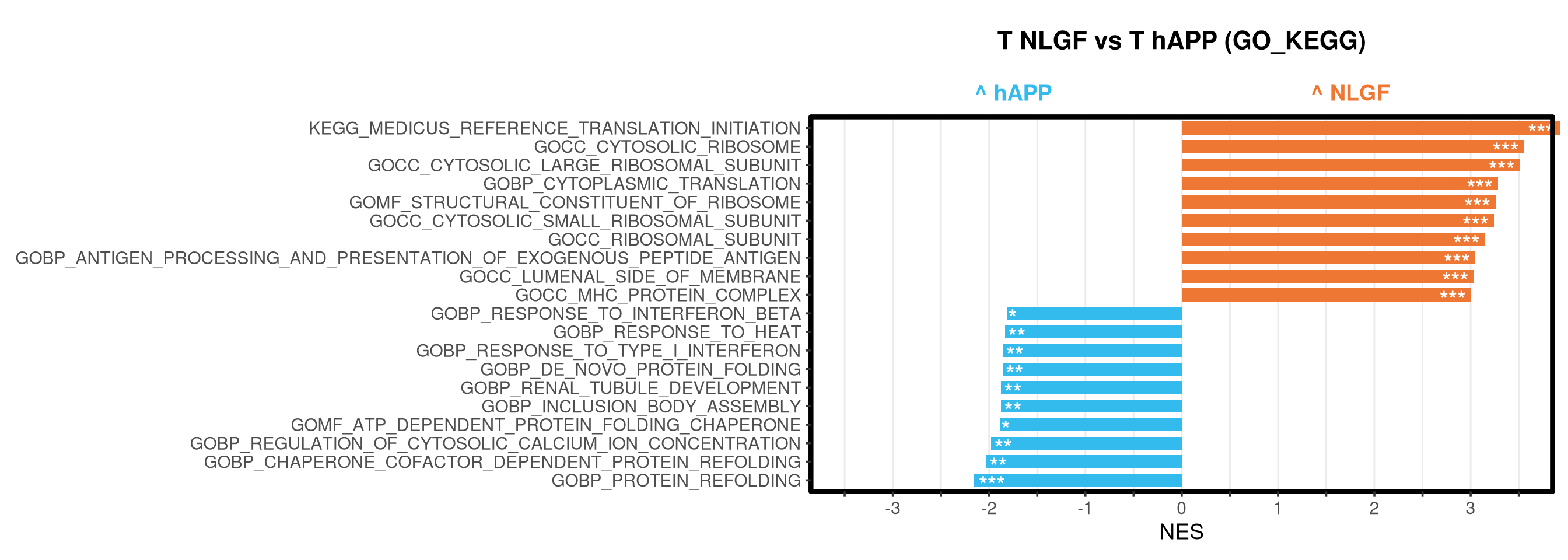
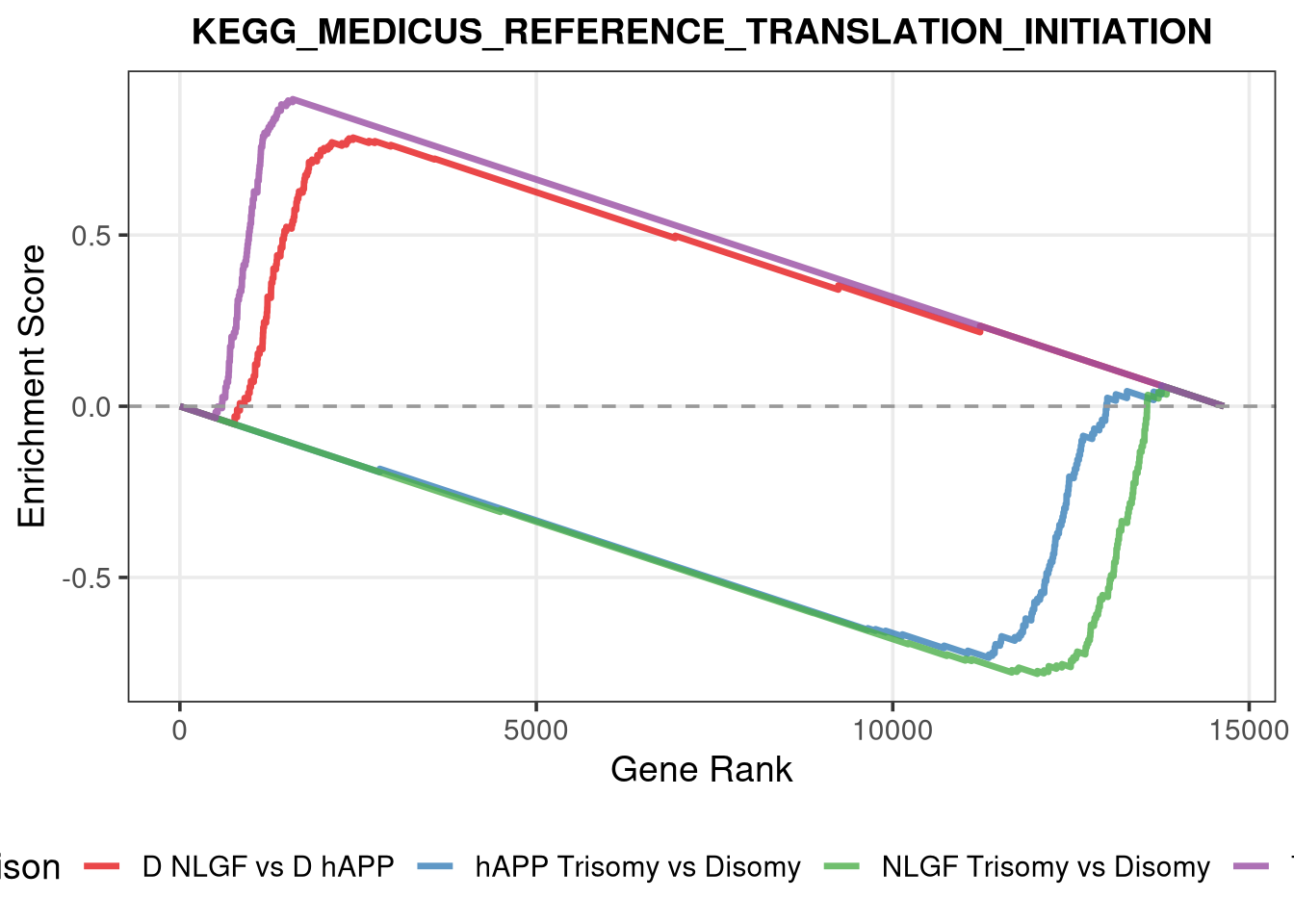
<- function (ranks,group_A_label = "Group A" ,group_B_label = "Group B" ,color_A = "#EE7733" ,color_B = "#33BBEE" ) {message (glue ("Running GSEA | Set: {geneset_name} | Comp: {comparison_name}" ))<- file.path (base_objects_dir, "GSEA_Analysis" , comparison_name)<- file.path (base_graphs_dir, "GSEA_Analysis" , comparison_name)dir.create (current_objects_dir, recursive = TRUE , showWarnings = FALSE )dir.create (current_graphs_dir, recursive = TRUE , showWarnings = FALSE )set.seed (42 )<- suppressMessages (fgsea (pathways = pathways,stats = ranks,minSize = 15 ,maxSize = 3000 ,nproc = 1 ,BPPARAM = BiocParallel:: SerialParam (progressbar = FALSE )if (nrow (fgsea_res) == 0 ) {warning (glue ("No significant pathways for {geneset_name} in {comparison_name}" ))return (NULL )<- fgsea_res %>% as_tibble () %>% mutate (leadingEdge = sapply (leadingEdge, paste, collapse = "," )) %>% mutate (pathway = gsub ("_geneset" , "" , pathway)) %>% arrange (padj)<- glue ("Table_{geneset_name}.csv" )write.csv (fgsea_res_tidy, file.path (current_objects_dir, csv_name), row.names = FALSE )<- fgsea_res_tidy %>% arrange (desc (NES)) %>% slice_head (n = 10 ) %>% bind_rows (fgsea_res_tidy %>% arrange (NES) %>% slice_head (n = 10 )) %>% distinct (pathway, .keep_all = TRUE ) %>% mutate (sig_label = case_when (< 0.001 ~ "***" ,< 0.01 ~ "**" ,< 0.05 ~ "*" ,TRUE ~ "" fill_group = ifelse (NES > 0 , group_A_label, group_B_label),pathway = factor (pathway, levels = unique (pathway[order (NES)]))if (nrow (top_gsea) == 0 ) return (fgsea_res_tidy)<- 3.5 <- glue ("{sub('_.*', '', comparison_name)} ({geneset_name})" )<- ggplot (top_gsea, aes (x = NES, y = pathway, fill = fill_group)) + geom_col (width = 0.7 ) + geom_text (aes (label = sig_label, hjust = ifelse (NES > 0 , 1.2 , - 0.2 )),color = "white" , size = 5 , vjust = 0.75 , fontface = "bold" ) + scale_fill_manual (values = setNames (c (color_A, color_B), c (group_A_label, group_B_label))) + scale_x_continuous (breaks = seq (- axis_zoom, axis_zoom, 0.5 ),labels = function (x) ifelse (x %% 1 == 0 , x, "" )+ scale_y_discrete (labels = function (x) str_wrap (x, width = 40 )) + labs (x = "NES" , y = NULL , title = plot_title) + theme_bw (base_size = 14 ) + theme (panel.grid.major.x = element_line (color = "grey92" , linewidth = 0.5 ),panel.grid.major.y = element_blank (),panel.grid.minor = element_blank (),panel.border = element_rect (colour = "black" , fill = NA , linewidth = 1.5 ),legend.position = "none" ,plot.title = element_text (hjust = 0.5 , face = "bold" , size = 16 , margin = margin (b = 40 )),plot.margin = margin (t = 20 , r = 10 , b = 10 , l = 10 , unit = "pt" )+ annotate ("text" , x = - axis_zoom/ 2 , y = Inf ,label = glue ("^ {group_B_label}" ), color = color_B, size = 5 , fontface = "bold" , vjust = - 1.0 ) + annotate ("text" , x = axis_zoom/ 2 , y = Inf ,label = glue ("^ {group_A_label}" ), color = color_A, size = 5 , fontface = "bold" , vjust = - 1.0 ) + coord_cartesian (xlim = c (- axis_zoom, axis_zoom), clip = "off" )<- glue ("Barplot_{geneset_name}.pdf" )<- 2 + (nrow (top_gsea) * 0.3 )<- max (nchar (as.character (top_gsea$ pathway)))<- 5 + (max_len * 0.12 )ggsave (filename = file.path (current_graphs_dir, plot_name),plot = p,width = dynamic_width,height = dynamic_height,limitsize = FALSE )print (p)return (fgsea_res_tidy)