Code
library(Seurat)
library(glmmTMB)
library(qs2)
library(dplyr)
library(tidyr)
library(ggplot2)
library(ggrepel)Visualises differential expression results from the glmmTMB model fitted in scripts/glmmTMB.qmd. Produces an interaction logFC scatter plot, per-gene violin and pseudobulk boxplots, eCDF plots, and ISG/top-gene summaries.
Inputs: output/run1/objects/glmmTMB_with_interactions_corrected.qs2, output/run1/objects/prelabelled_integrated_rpca.qs2 Outputs: PDF figures written to output/run1/graphs/
library(Seurat)
library(glmmTMB)
library(qs2)
library(dplyr)
library(tidyr)
library(ggplot2)
library(ggrepel)run_num <- "run1"
out_dir <- file.path("output", run_num)
graphs_dir <- file.path(out_dir, "graphs")
objects_dir <- file.path(out_dir, "objects")
dir.create(graphs_dir, recursive = TRUE, showWarnings = FALSE)
dir.create(objects_dir, recursive = TRUE, showWarnings = FALSE)required_inputs <- c(
file.path(objects_dir, "glmmTMB_with_interactions_corrected.qs2"),
file.path(objects_dir, "prelabelled_integrated_rpca.qs2")
)
missing <- required_inputs[!file.exists(required_inputs)]
if (length(missing) > 0) {
message("Required inputs not found:\n", paste(" -", missing, collapse = "\n"),
"\nRun scripts/glmmTMB.qmd and seurat_trisomy_v1/v2.qmd first.")
knitr::knit_exit()
}
final_df <- qs_read(required_inputs[1])
seurat_obj <- qs_read(required_inputs[2])plot_data <- final_df %>%
# 1. Keep only the rows where we are looking at the Ploidy effect (A vs B)
# and ensure Background is one of our two target groups.
filter(contrast == "A - B", !is.na(Background)) %>%
# 2. Select only the necessary columns.
# Important: Interaction_FDR must be the same for both rows of a gene
# or you'll get duplicates again.
select(Gene, Background, estimate, Interaction_FDR) %>%
# 3. Pivot: this should now have 1 row per Gene
pivot_wider(names_from = Background, values_from = estimate) %>%
# 4. Create labeling logic
mutate(
Significance = ifelse(Interaction_FDR < 0.05, "Significant Interaction", "Not Significant"),
Label = ifelse(Interaction_FDR < 0.05, Gene, NA)
)p <- ggplot(plot_data, aes(x = NLGF, y = hAPP)) +
# Draw the non-significant genes first so they fall in the background
geom_point(data = filter(plot_data, Significance == "Not Significant"),
color = "grey80", alpha = 0.5, size = 1.5) +
# Draw your 23 significant genes on top in a bold color
geom_point(data = filter(plot_data, Significance == "Significant Interaction"),
color = "red", alpha = 0.9, size = 2.5) +
# Add the "Zero" crosshairs to divide it into quadrants
geom_hline(yintercept = 0, linetype = "dotted", color = "black") +
geom_vline(xintercept = 0, linetype = "dotted", color = "black") +
# Add the diagonal "Line of No Interaction" (Slope = 1, Intercept = 0)
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "darkblue", linewidth = 0.8) +
# Add the repelling text labels for the 23 genes
geom_text_repel(aes(label = Label),
color = "black",
size = 4,
fontface = "bold",
box.padding = 0.5,
max.overlaps = Inf) + # Ensures it tries to plot all 23 labels
# Aesthetics
theme_minimal() +
labs(
title = "Interaction Landscape: Trisomy Effect by Plaque Background",
x = "Log2 Fold Change (Trisomic vs Disomic in NLGF)",
y = "Log2 Fold Change (Trisomic vs Disomic in hAPP)"
) +
theme(
plot.title = element_text(face = "bold", size = 14),
axis.title = element_text(face = "bold")
)
print(p)Warning: Removed 14629 rows containing missing values or values outside the scale range
(`geom_text_repel()`).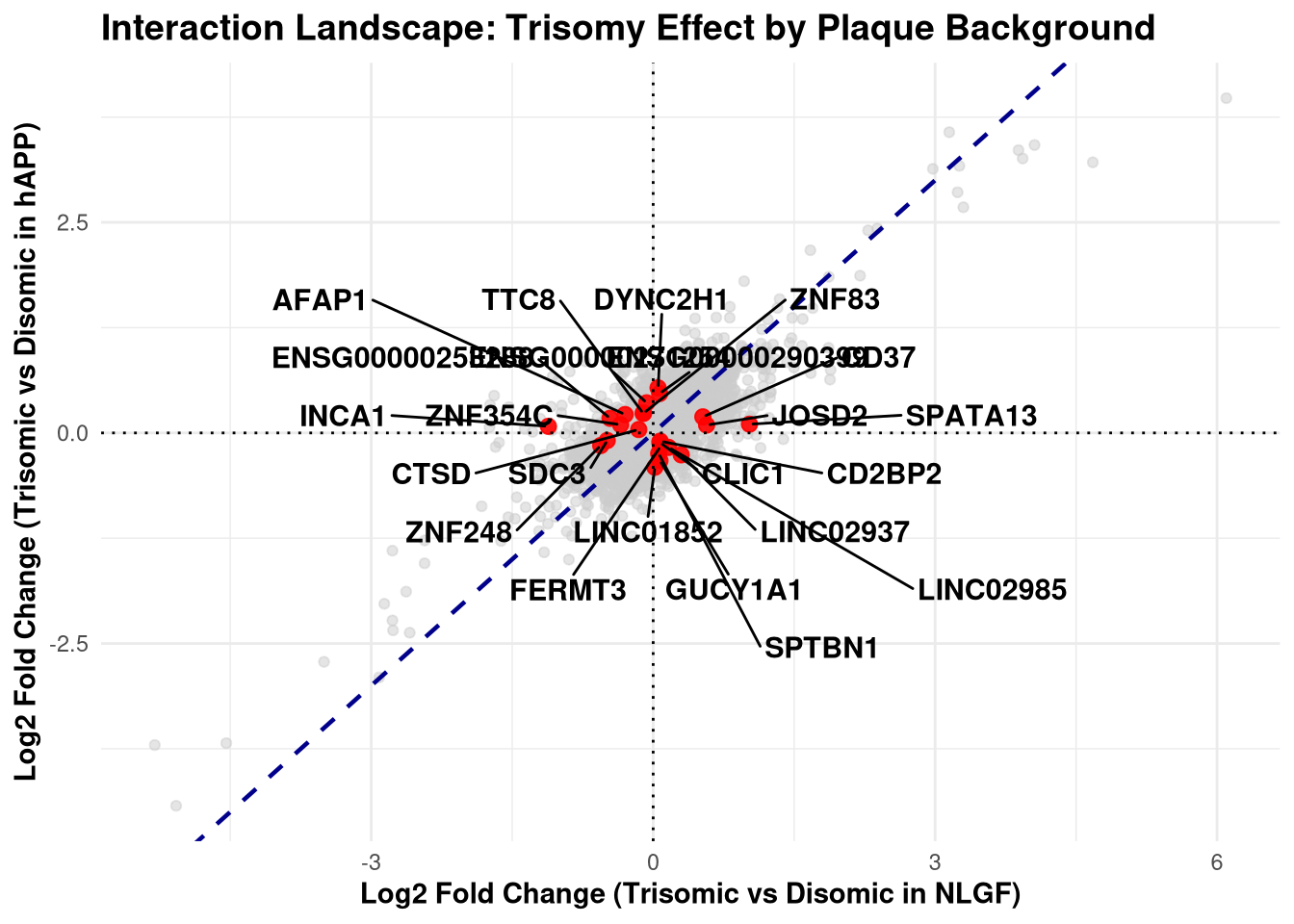
ggsave(
file.path(graphs_dir, "interaction_logFC_scatterplot.pdf"),
plot = p,
width = 12,
height = 6,
units = "in"
)Warning: Removed 14629 rows containing missing values or values outside the scale range
(`geom_text_repel()`).#Extract signficant interaction genes from plot data
sig_genes <- plot_data %>%
filter(Significance == "Significant Interaction") %>%
pull(Gene)
sig_genes [1] "AFAP1" "CD2BP2" "CD37" "CLIC1"
[5] "CTSD" "DYNC2H1" "ENSG00000253288" "ENSG00000271254"
[9] "ENSG00000290399" "FERMT3" "GUCY1A1" "INCA1"
[13] "JOSD2" "LINC01852" "LINC02937" "LINC02985"
[17] "SDC3" "SPATA13" "SPTBN1" "TTC8"
[21] "ZNF248" "ZNF354C" "ZNF83" #Pull raw counts for these genes
expr_matrix <- GetAssayData(seurat_obj, assay = "SCT", layer = "data")[sig_genes, ]
expr_df <- as.data.frame(t(as.matrix(expr_matrix)))
expr_df$CellID <- rownames(expr_df)
expr_df$Ploidy <- factor(seurat_obj@meta.data$microglia, levels = c("B", "A"))
expr_df$Background <- factor(seurat_obj@meta.data$app_genotype, levels = c("NLGF", "hAPP"))
expr_df$Mouse <- seurat_obj@meta.data$orig.ident
expr_df$Condition <- factor(seurat_obj@meta.data$group_id)
# Pivot to "Long" format for ggplot2
expr_long <- expr_df %>%
pivot_longer(cols = any_of(sig_genes), names_to = "Gene", values_to = "Expression")
pdf_file <- file.path(graphs_dir, "Interaction_Genes_Raw_Violins.pdf")
pdf(pdf_file, width = 8, height = 6)
for (gene in sig_genes) {
# Filter data for the current gene
gene_data <- expr_long %>% filter(Gene == gene)
# Create the plot
p <- ggplot(gene_data, aes(x = Condition, y = Expression, fill = Background)) +
# scale="width" ensures all violins are the same width regardless of cell count
geom_violin(scale = "width", alpha = 0.7, color = "black") +
# Add a point for the mean expression to help guide the eye
stat_summary(fun = mean, geom = "point", shape = 23, size = 3, fill = "white") +
scale_fill_manual(values = c("NLGF" = "#4C72B0", "hAPP" = "#C44E52")) +
theme_minimal(base_size = 14) +
labs(
title = paste("Expression of", gene),
subtitle = "Significant Ploidy * Background Interaction",
x = "Condition",
y = "Raw Counts"
) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
axis.text.x = element_text(face = "bold"),
legend.position = "none" # We don't need a legend since the X-axis is labeled
)
print(p) # Print each plot as a new page in the PDF
}
dev.off() # Close and save the PDFpng
2 cat("Saved 23 violin plots to:", pdf_file, "\n")Saved 23 violin plots to: output/run1/graphs/Interaction_Genes_Raw_Violins.pdf p_dot <- DotPlot(seurat_obj,
features = sig_genes,
group.by = "group_id",
assay = "SCT") +
# Improve the aesthetics for a large number of genes
theme(axis.text.x = element_text(angle = 45, hjust = 1, face = "bold.italic"),
axis.text.y = element_text(face = "bold", size = 12),
plot.title = element_text(face = "bold", hjust = 0.5)) +
# Custom colors: Low expression is light grey, high expression is dark red
scale_color_gradient(low = "lightgrey", high = "#C44E52") +
labs(title = "Significant Ploidy * Background Interaction Genes",
x = "Gene",
y = "Condition")Warning: Scaling data with a low number of groups may produce misleading
resultsScale for colour is already present.
Adding another scale for colour, which will replace the existing scale.# 3. SAVE TO PDF
# Because you have 23 genes, a wider PDF (e.g., width = 12) prevents the gene names from squishing
dp_file <- file.path(graphs_dir, "Interaction_Genes_DotPlot.pdf")
pdf(dp_file, width = 12, height = 5)
print(p_dot)
dev.off()png
2 cat("DotPlot saved to:", dp_file, "\n")DotPlot saved to: output/run1/graphs/Interaction_Genes_DotPlot.pdf # Collapse the single cells down to one average value per mouse
pb_long <- expr_long %>%
# We group by Mouse, Condition, and Background so we keep those labels for plotting
group_by(Mouse, Condition, Background, Gene) %>%
summarise(Pseudobulk_Expression = mean(Expression), .groups = "drop")
# 4. PLOT AND SAVE TO PDF
pdf_file <- file.path(graphs_dir, "Interaction_Genes_Pseudobulk.pdf")
pdf(pdf_file, width = 8, height = 6)
for (gene in sig_genes) {
# Filter data for the current gene
gene_pb <- pb_long %>% filter(Gene == gene)
# Create the boxplot
p <- ggplot(gene_pb, aes(x = Condition, y = Pseudobulk_Expression, fill = Background)) +
# Add the boxplot structure (hide outliers since we will draw all mice manually)
geom_boxplot(alpha = 0.5, outlier.shape = NA) +
# Draw every single mouse as a distinct dot (jittered slightly so they don't overlap)
geom_jitter(width = 0.2, size = 3, shape = 21, color = "black", stroke = 1) +
# Use your defined colors for the background
scale_fill_manual(values = c("NLGF" = "#4C72B0", "hAPP" = "#C44E52")) +
theme_minimal(base_size = 14) +
labs(
title = paste(gene, "- Pseudobulk Expression"),
subtitle = "Each dot represents the average normalized expression of one mouse",
x = "Condition",
y = "Mean SCT Normalized Expression"
) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
# Angle the X-axis labels in case your group_id names are long
axis.text.x = element_text(face = "bold", angle = 45, hjust = 1),
legend.position = "none" # Hide legend since X-axis/colors explain it
)
print(p) # Print each plot as a new page in the PDF
}
dev.off() # Close and save the PDFpng
2 cat("Saved pseudobulked boxplots to:", pdf_file, "\n")Saved pseudobulked boxplots to: output/run1/graphs/Interaction_Genes_Pseudobulk.pdf ecdf_pdf_file <- file.path(graphs_dir, "Interaction_Genes_NonZero_eCDF_with_Proportions.pdf")
pdf(ecdf_pdf_file, width = 10, height = 7)
for (gene in sig_genes) {
# 1. Get data for the specific gene
gene_data_all <- expr_long %>% filter(Gene == gene)
# 2. Calculate the proportion of non-zero cells per group
stats_summary <- gene_data_all %>%
group_by(Condition) %>%
summarise(
total_cells = n(),
non_zero_cells = sum(Expression > 0),
prop_expressing = (non_zero_cells / total_cells) * 100,
.groups = "drop"
) %>%
mutate(label = paste0(Condition, ": ", round(prop_expressing, 1), "%"))
# Create a string to display as a subtitle
prop_text <- paste("Expressing Cells % -", paste(stats_summary$label, collapse = " | "))
# 3. Filter for non-zero cells for the eCDF
gene_data_nonzero <- gene_data_all %>% filter(Expression > 0)
if(nrow(gene_data_nonzero) < 5) {
message("Skipping ", gene, " - too few expressing cells.")
next
}
# 4. Create the plot
p_ecdf <- ggplot(gene_data_nonzero, aes(x = Expression, color = Condition)) +
stat_ecdf(geom = "step", linewidth = 1.2) +
# We use a log scale if the data is highly skewed, but SCT is usually okay on linear
theme_minimal(base_size = 12) +
labs(
title = paste(gene, "- Non-Zero Distribution"),
subtitle = prop_text,
x = "SCT Normalized Expression (Non-Zero Only)",
y = "Cumulative Fraction of Expressing Cells",
color = "Condition"
) +
theme(
plot.title = element_text(face = "bold", size = 16),
plot.subtitle = element_text(size = 9, color = "blue4"),
legend.position = "bottom",
panel.grid.minor = element_blank()
)
print(p_ecdf)
}
dev.off()png
2 cat("Saved annotated eCDF plots to:", ecdf_pdf_file, "\n")Saved annotated eCDF plots to: output/run1/graphs/Interaction_Genes_NonZero_eCDF_with_Proportions.pdf # Updated file name to reflect all cells
ecdf_pdf_file <- file.path(graphs_dir, "Interaction_Genes_AllCells_eCDF_with_Proportions.pdf")
pdf(ecdf_pdf_file, width = 10, height = 7)
for (gene in sig_genes) {
# 1. Get data for the specific gene (includes all zeros)
gene_data_all <- expr_long %>% filter(Gene == gene)
# 2. Calculate the proportion of non-zero cells per group
stats_summary <- gene_data_all %>%
group_by(Condition) %>%
summarise(
total_cells = n(),
non_zero_cells = sum(Expression > 0),
prop_expressing = (non_zero_cells / total_cells) * 100,
.groups = "drop"
) %>%
mutate(label = paste0(Condition, ": ", round(prop_expressing, 1), "%"))
# Create a string to display as a subtitle
prop_text <- paste("Expressing Cells % -", paste(stats_summary$label, collapse = " | "))
# 3. Check to ensure we have enough total cells to plot
if(nrow(gene_data_all) < 5) {
message("Skipping ", gene, " - too few total cells.")
next
}
# 4. Create the plot using gene_data_all instead of gene_data_nonzero
p_ecdf <- ggplot(gene_data_all, aes(x = Expression, color = Condition)) +
stat_ecdf(geom = "step", linewidth = 1.2) +
theme_minimal(base_size = 12) +
labs(
title = paste(gene, "- All Cells Distribution"),
subtitle = prop_text,
x = "SCT Normalized Expression (All Cells)",
y = "Cumulative Fraction of Total Cells",
color = "Condition"
) +
theme(
plot.title = element_text(face = "bold", size = 16),
plot.subtitle = element_text(size = 9, color = "blue4"),
legend.position = "bottom",
panel.grid.minor = element_blank()
)
print(p_ecdf)
}
dev.off()png
2 cat("Saved annotated eCDF plots to:", ecdf_pdf_file, "\n")Saved annotated eCDF plots to: output/run1/graphs/Interaction_Genes_AllCells_eCDF_with_Proportions.pdf isg_genes <- c("IFI27", "IFI44L", "IFIT1", "IFIT2", "IFIT3",
"MX1", "MX2", "OAS1", "ISG15", "USP18",
"RSAD2", "LY6E", "DDX60", "TRIM25", "STAT1", "IRF7")
isg_nlgf <- final_df %>%
filter(Background == "NLGF" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(Gene %in% isg_genes) %>%
slice_head(n = 10)
isg_happ <- final_df %>%
filter(Background == "hAPP" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(Gene %in% isg_genes) %>%
slice_head(n = 10)
expr_matrix <- GetAssayData(seurat_obj, assay = "SCT", layer = "data")[isg_genes, ]
expr_df <- as.data.frame(t(as.matrix(expr_matrix)))
expr_df$CellID <- rownames(expr_df)
expr_df$Ploidy <- factor(seurat_obj@meta.data$microglia, levels = c("B", "A"))
expr_df$Background <- factor(seurat_obj@meta.data$app_genotype, levels = c("NLGF", "hAPP"))
expr_df$Mouse <- seurat_obj@meta.data$orig.ident
expr_df$Condition <- factor(seurat_obj@meta.data$group_id)
expr_long <- expr_df %>%
pivot_longer(cols = any_of(isg_genes), names_to = "Gene", values_to = "Expression")
# Collapse the single cells down to one average value per mouse
pb_long <- expr_long %>%
# We group by Mouse, Condition, and Background so we keep those labels for plotting
group_by(Mouse, Condition, Background, Gene) %>%
summarise(Pseudobulk_Expression = mean(Expression), .groups = "drop")
# 4. PLOT AND SAVE TO PDF
pdf_file <- file.path(graphs_dir, "ISG_genes_Pseudobulk_glmmtmb.pdf")
pdf(pdf_file, width = 8, height = 6)
for (gene in isg_genes) {
# Filter data for the current gene
gene_pb <- pb_long %>% filter(Gene == gene)
# Extract the FDR values for BOTH backgrounds from final_df
nlgf_fdr_raw <- final_df %>% filter(Gene == gene & Background == "NLGF" & contrast == "A - B") %>% pull(FDR)
happ_fdr_raw <- final_df %>% filter(Gene == gene & Background == "hAPP" & contrast == "A - B") %>% pull(FDR)
# Format cleanly, handling potential missing values gracefully
fmt_nlgf <- ifelse(length(nlgf_fdr_raw) > 0, signif(nlgf_fdr_raw, digits = 3), "N/A")
fmt_happ <- ifelse(length(happ_fdr_raw) > 0, signif(happ_fdr_raw, digits = 3), "N/A")
# Create the boxplot
p <- ggplot(gene_pb, aes(x = Condition, y = Pseudobulk_Expression, fill = Background)) +
# Add the boxplot structure (hide outliers since we will draw all mice manually)
geom_boxplot(alpha = 0.5, outlier.shape = NA) +
# Draw every single mouse as a distinct dot (jittered slightly so they don't overlap)
geom_jitter(width = 0.2, size = 3, shape = 21, color = "black", stroke = 1) +
# Use your defined colors for the background
scale_fill_manual(values = c("NLGF" = "#4C72B0", "hAPP" = "#C44E52")) +
theme_minimal(base_size = 14) +
labs(
title = paste(gene, "- Pseudobulk Expression"),
subtitle = paste0("FDR (NLGF): ", fmt_nlgf, " | FDR (hAPP): ", fmt_happ, "\nEach dot = average expression of one mouse"),
x = "Condition",
y = "Mean SCT Normalized Expression"
) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
# Angle the X-axis labels in case your group_id names are long
axis.text.x = element_text(face = "bold", angle = 45, hjust = 1),
legend.position = "none" # Hide legend since X-axis/colors explain it
)
print(p) # Print each plot as a new page in the PDF
}
dev.off() # Close and save the PDFpng
2 cat("Saved pseudobulked boxplots to:", pdf_file, "\n")Saved pseudobulked boxplots to: output/run1/graphs/ISG_genes_Pseudobulk_glmmtmb.pdf pb_long <- expr_long %>%
# We group by Mouse, Condition, and Background so we keep those labels for plotting
group_by(Mouse, Condition, Background, Gene) %>%
summarise(Pseudobulk_Expression = mean(Expression), .groups = "drop")
top10_sig <- final_df %>%
filter(Background == "NLGF" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(estimate > 0) %>%
slice_head(n = 10)
bot10_sig <- final_df %>%
filter(Background == "NLGF" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(estimate < 0) %>%
slice_head(n = 10)
top_bottom_res <- bind_rows(top10_sig, bot10_sig)
target_genes <- top_bottom_res$Gene
pdf_file <- file.path(graphs_dir, "ISG_genes_top_bottom_nlgf_a_vs_b.pdf")
pdf(pdf_file, width = 8, height = 6)
for (gene in target_genes) {
# Filter data for the current gene
gene_pb <- pb_long %>% filter(Gene == gene)
# Extract the FDR value for this specific gene from the glmmTMB results table
# Using signif() to format it cleanly (e.g., 0.000123 -> 1.23e-04)
gene_fdr <- top_bottom_res %>% filter(Gene == gene) %>% pull(FDR)
formatted_fdr <- signif(gene_fdr, digits = 3)
# Create the boxplot
p <- ggplot(gene_pb, aes(x = Condition, y = Pseudobulk_Expression, fill = Background)) +
# Add the boxplot structure (hide outliers since we will draw all mice manually)
geom_boxplot(alpha = 0.5, outlier.shape = NA) +
# Draw every single mouse as a distinct dot (jittered slightly so they don't overlap)
geom_jitter(width = 0.2, size = 3, shape = 21, color = "black", stroke = 1) +
# Use your defined colors for the background
scale_fill_manual(values = c("NLGF" = "#4C72B0", "hAPP" = "#C44E52")) +
theme_minimal(base_size = 14) +
labs(
title = paste(gene, "- Pseudobulk Expression"),
subtitle = paste0("glmmTMB FDR: ", formatted_fdr, " | Each dot = average expression of one mouse"),
x = "Condition",
y = "Mean SCT Normalized Expression"
) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
# Angle the X-axis labels in case your group_id names are long
axis.text.x = element_text(face = "bold", angle = 45, hjust = 1),
legend.position = "none" # Hide legend since X-axis/colors explain it
)
print(p) # Print each plot as a new page in the PDF
}Warning: No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.dev.off() # Close and save the PDFpng
2 cat("Saved pseudobulked boxplots to:", pdf_file, "\n")Saved pseudobulked boxplots to: output/run1/graphs/ISG_genes_top_bottom_nlgf_a_vs_b.pdf pb_long <- expr_long %>%
# We group by Mouse, Condition, and Background so we keep those labels for plotting
group_by(Mouse, Condition, Background, Gene) %>%
summarise(Pseudobulk_Expression = mean(Expression), .groups = "drop")
top10_sig <- final_df %>%
filter(Background == "hAPP" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(estimate > 0) %>%
slice_head(n = 10)
bot10_sig <- final_df %>%
filter(Background == "hAPP" & contrast == "A - B") %>%
arrange(FDR) %>%
filter(estimate < 0) %>%
slice_head(n = 10)
top_bottom_res <- bind_rows(top10_sig, bot10_sig)
target_genes <- top_bottom_res$Gene
pdf_file <- file.path(graphs_dir, "top10_bottom_happ_a_vs_b.pdf")
pdf(pdf_file, width = 8, height = 6)
for (gene in target_genes) {
# Filter data for the current gene
gene_pb <- pb_long %>% filter(Gene == gene)
# Extract the FDR value for this specific gene from the glmmTMB results table
# Using signif() to format it cleanly (e.g., 0.000123 -> 1.23e-04)
gene_fdr <- top_bottom_res %>% filter(Gene == gene) %>% pull(FDR)
formatted_fdr <- signif(gene_fdr, digits = 3)
# Create the boxplot
p <- ggplot(gene_pb, aes(x = Condition, y = Pseudobulk_Expression, fill = Background)) +
# Add the boxplot structure (hide outliers since we will draw all mice manually)
geom_boxplot(alpha = 0.5, outlier.shape = NA) +
# Draw every single mouse as a distinct dot (jittered slightly so they don't overlap)
geom_jitter(width = 0.2, size = 3, shape = 21, color = "black", stroke = 1) +
# Use your defined colors for the background
scale_fill_manual(values = c("NLGF" = "#4C72B0", "hAPP" = "#C44E52")) +
theme_minimal(base_size = 14) +
labs(
title = paste(gene, "- Pseudobulk Expression"),
subtitle = paste0("glmmTMB FDR: ", formatted_fdr, " | Each dot = average expression of one mouse"),
x = "Condition",
y = "Mean SCT Normalized Expression"
) +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
# Angle the X-axis labels in case your group_id names are long
axis.text.x = element_text(face = "bold", angle = 45, hjust = 1),
legend.position = "none" # Hide legend since X-axis/colors explain it
)
print(p) # Print each plot as a new page in the PDF
}Warning: No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.
No shared levels found between `names(values)` of the manual scale and the
data's fill values.dev.off() # Close and save the PDFpng
2 cat("Saved pseudobulked boxplots to:", pdf_file, "\n")Saved pseudobulked boxplots to: output/run1/graphs/top10_bottom_happ_a_vs_b.pdf