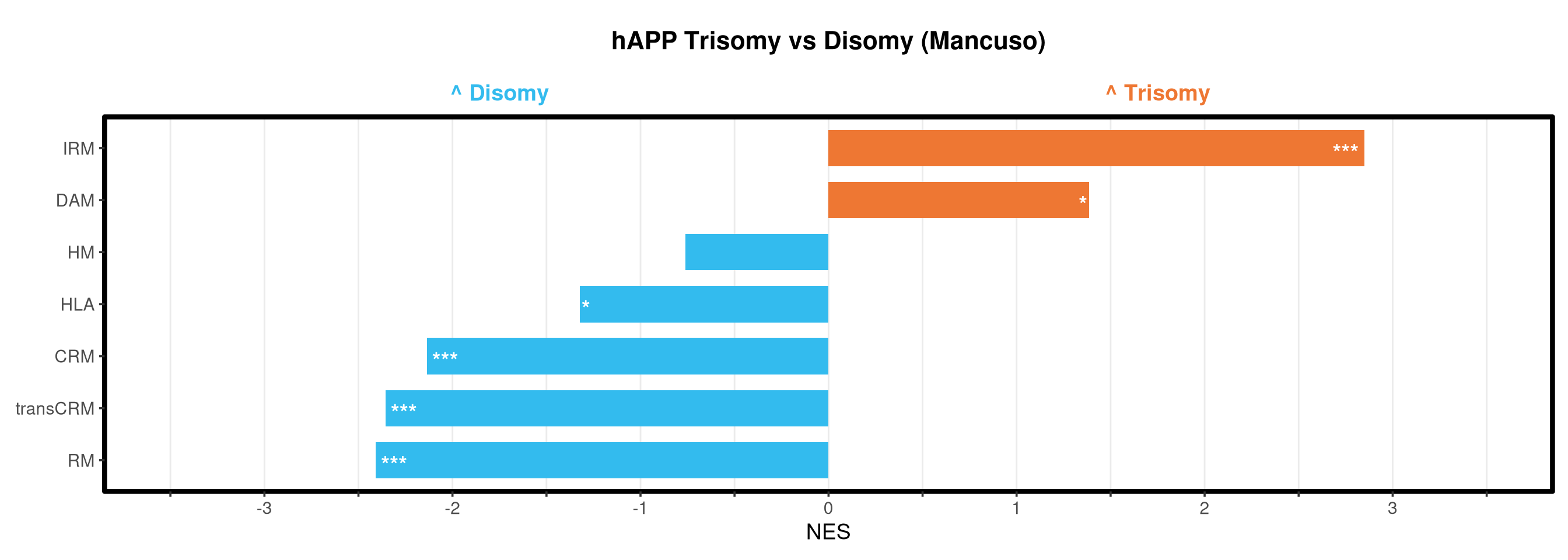
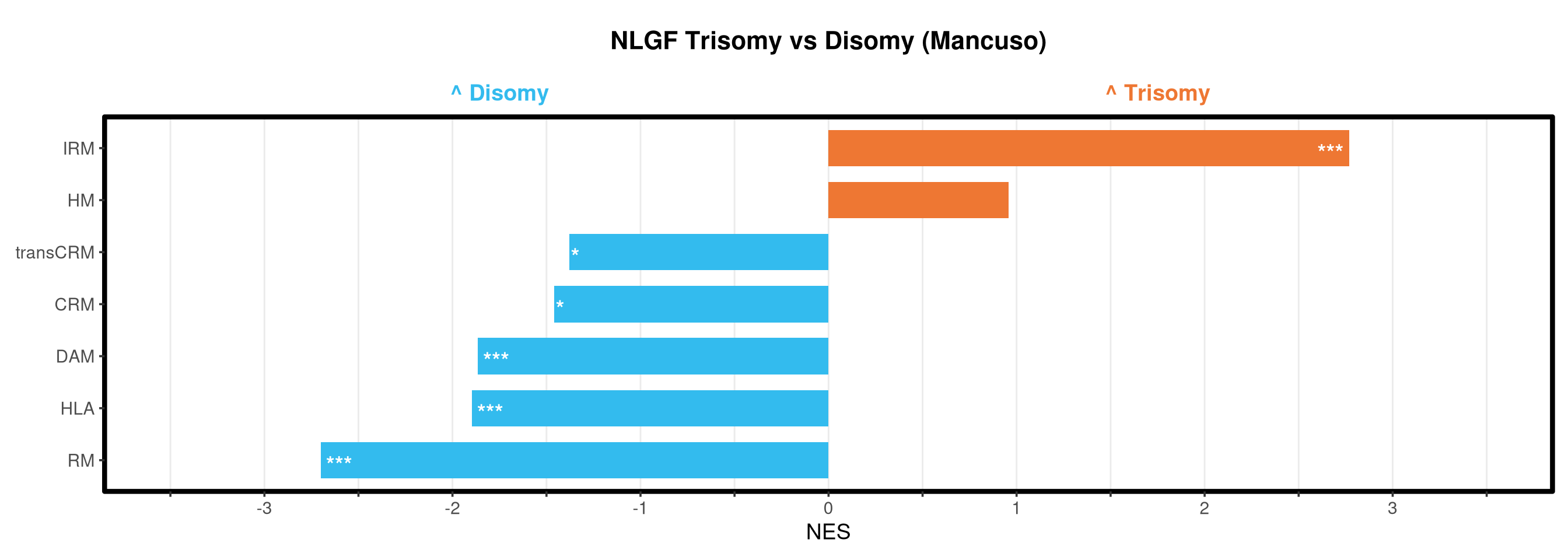
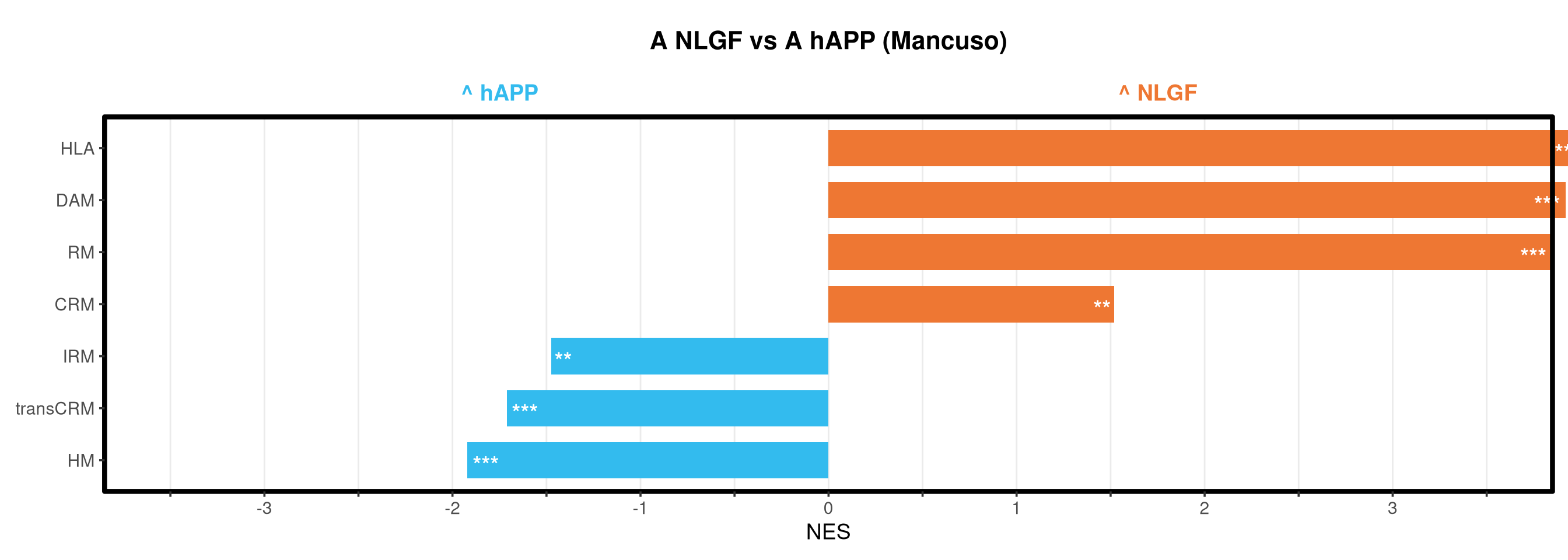
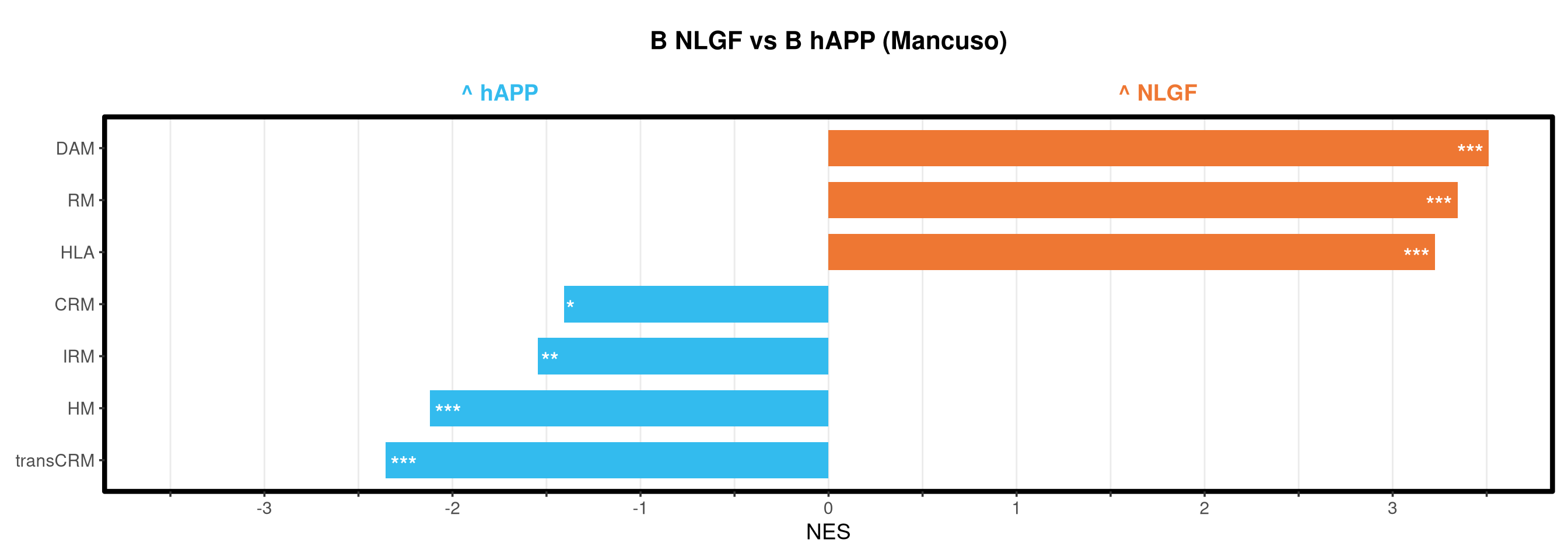
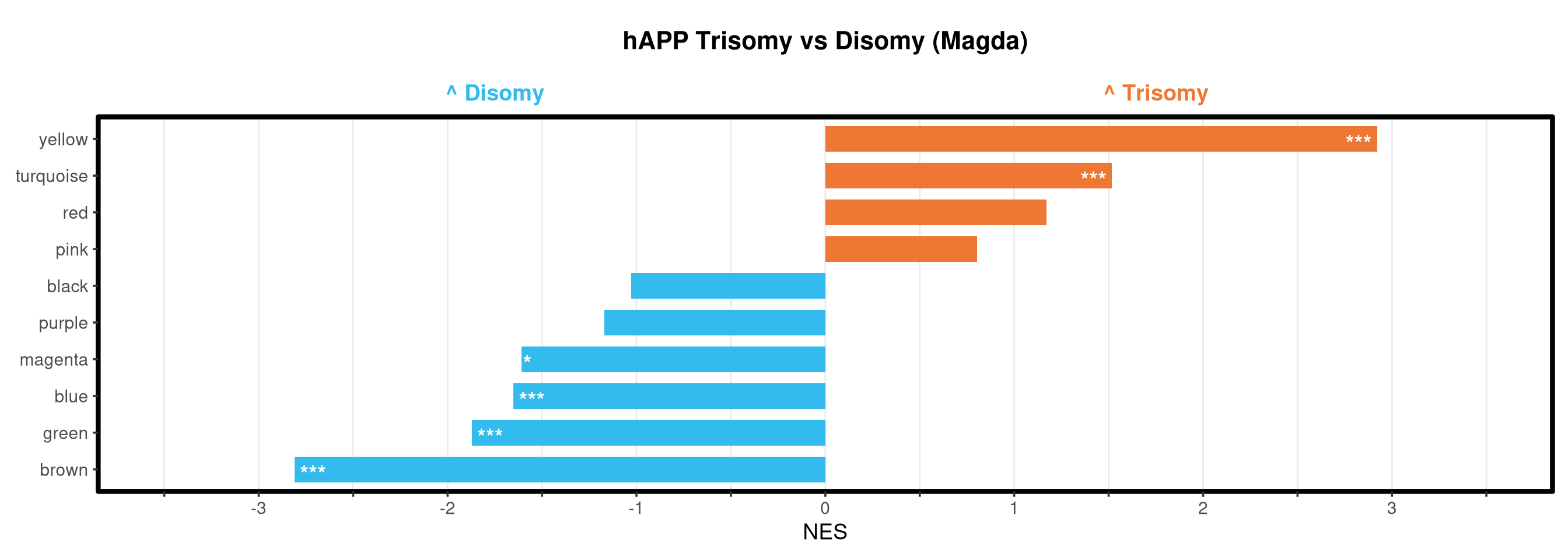
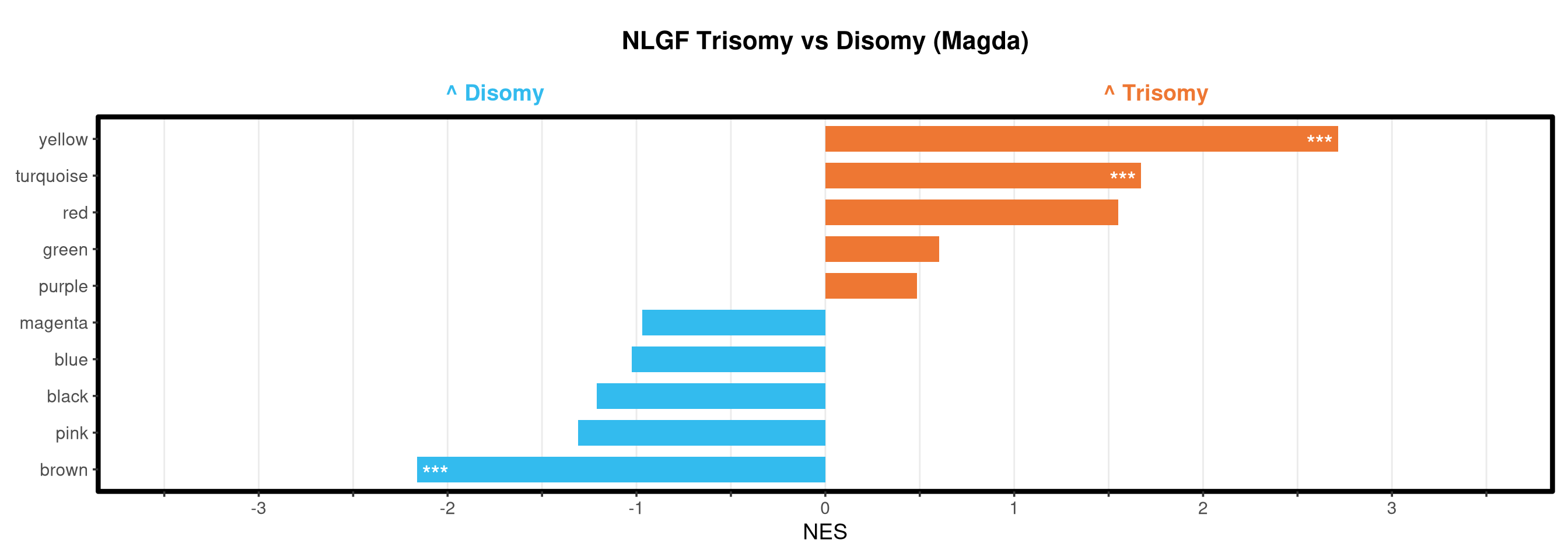
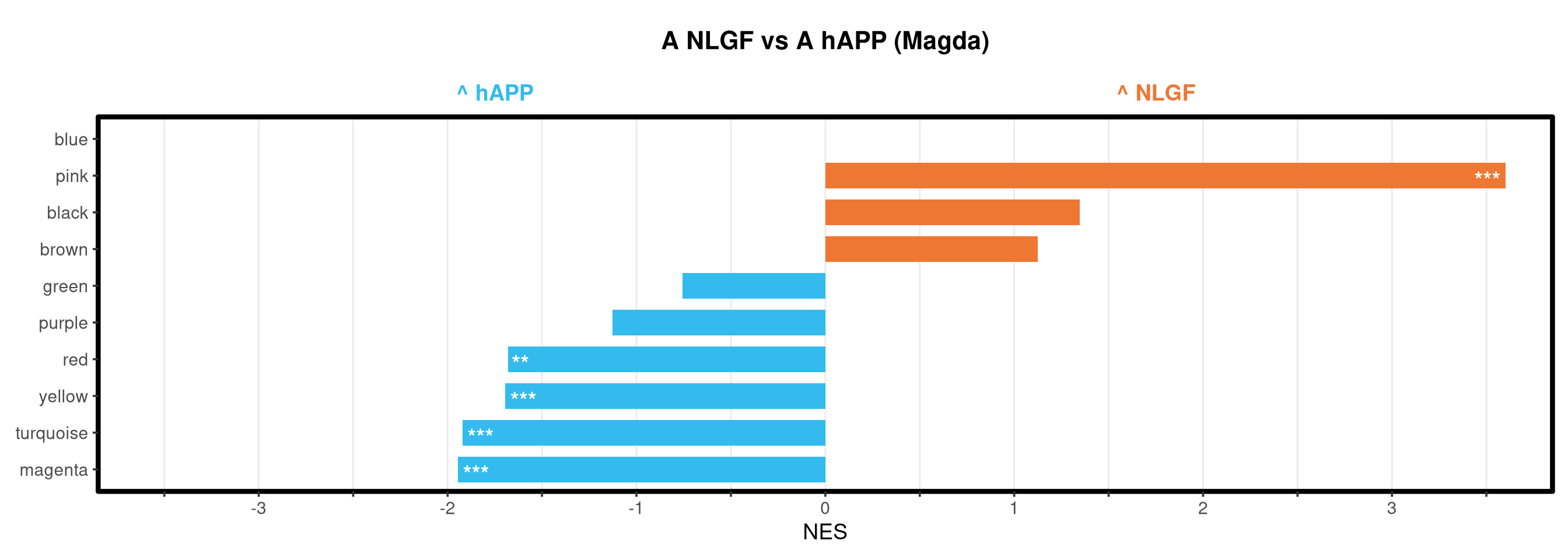
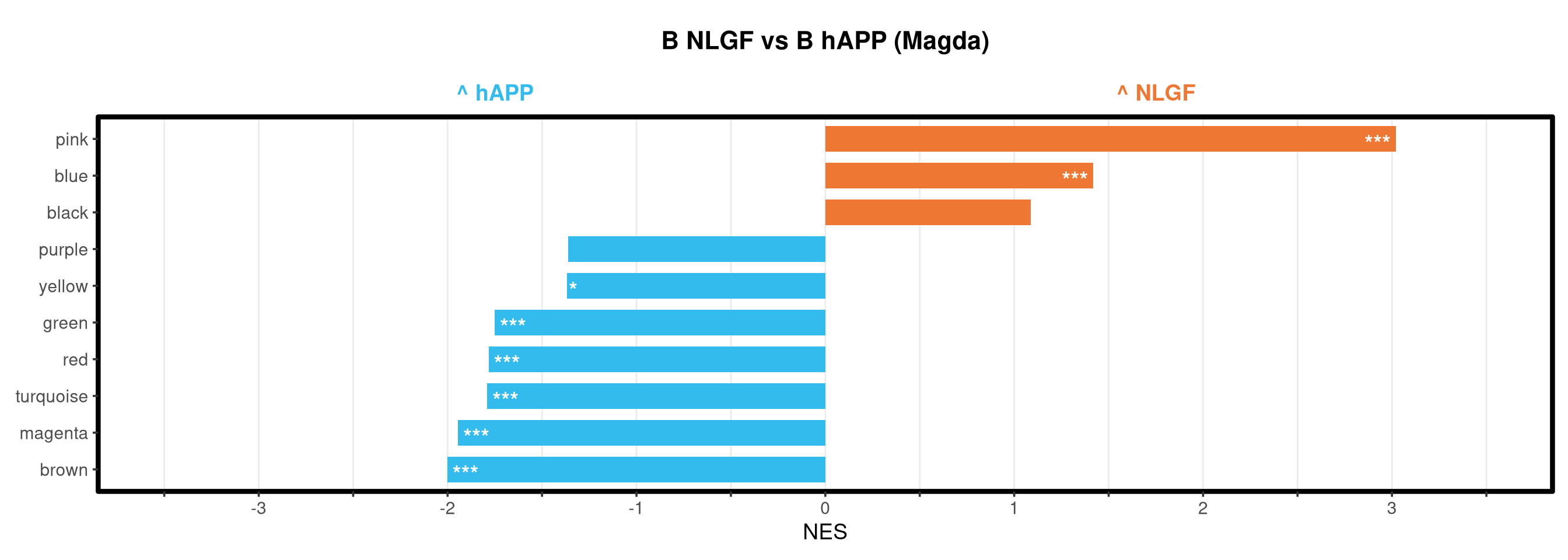
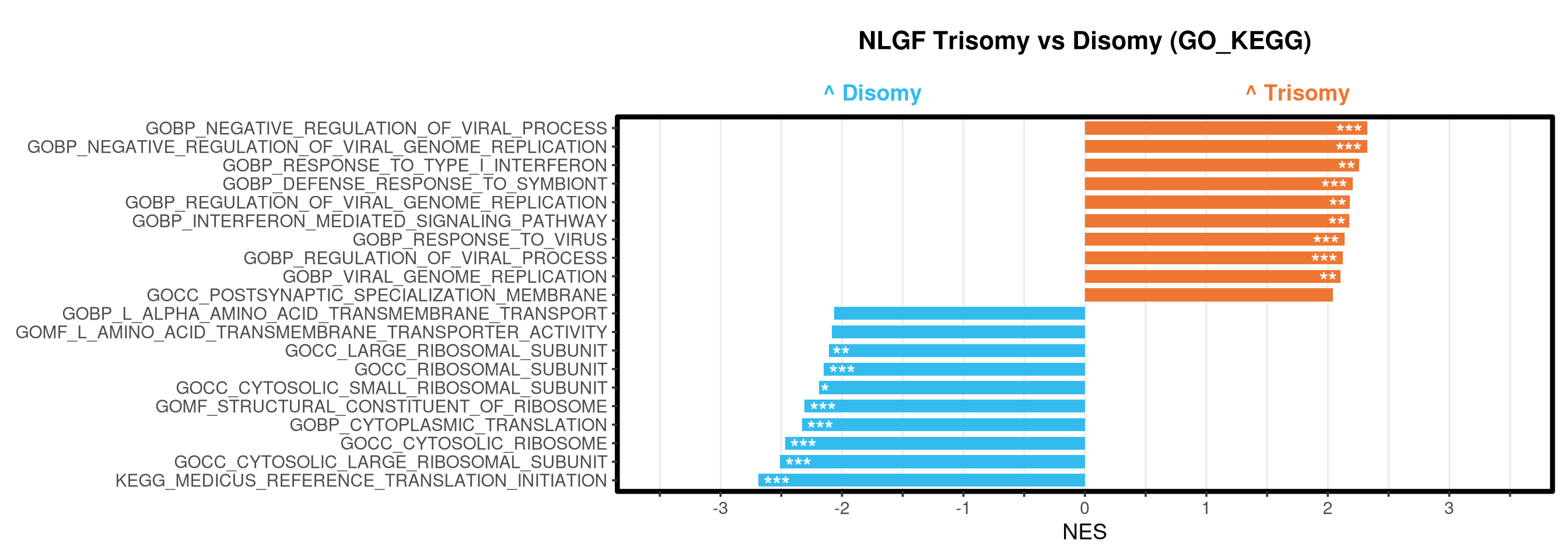
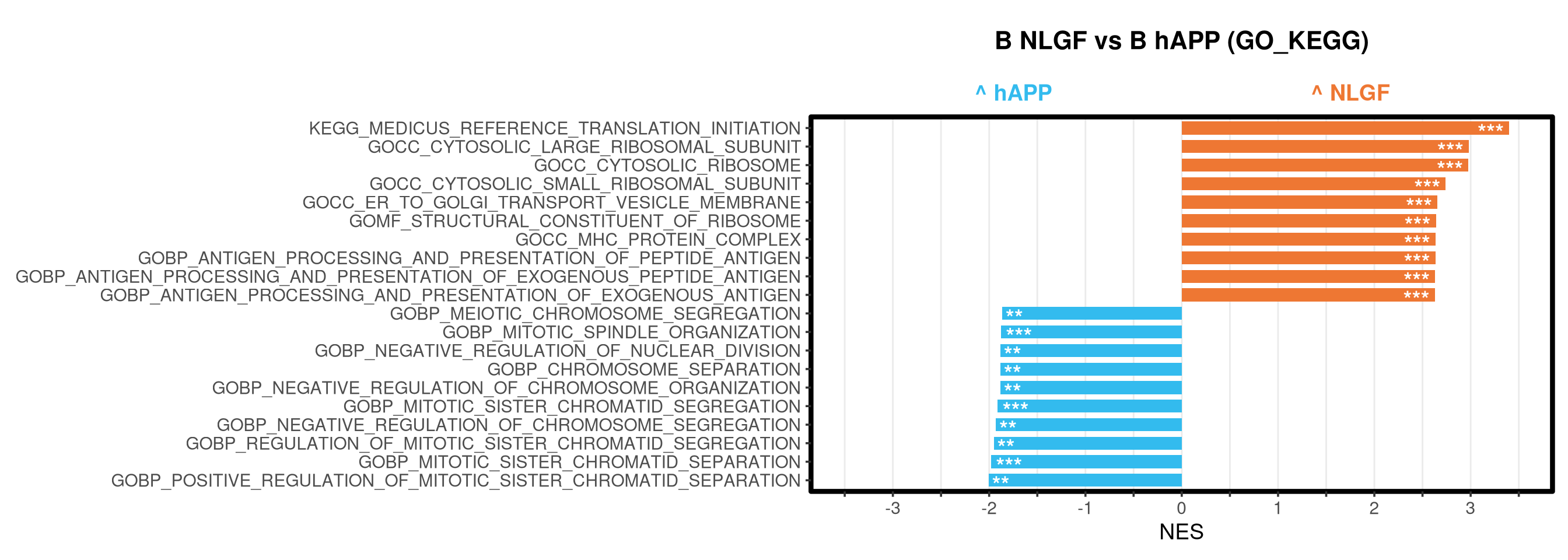
Runs gene set enrichment analysis (fgsea) on glmmTMB differential expression results across four contrasts: trisomy effect in hAPP and NLGF backgrounds, and background effect in trisomic and disomic cells. Gene sets include Mancuso 2024 signatures, custom module lists, and GO/KEGG pathways.
Inputs:output/run1/objects/glmmTMB_with_interactions_corrected.qs2; Mancuso geneset TXTs (../../Mancuso2024_genesets/); Magda modules Excel; GO/KEGG .gmtOutputs: CSV tables and barplot PDFs written to output/run1/glmmTMB/GSEA/
purrr::walk2(names(gene_sets_master), gene_sets_master, function(gset_name, gset_pathways) { purrr::walk2(comparisons_master, names(comparisons_master), function(ranks, comp_name) {# 1. Dynamically determine group labels based on the current comparison name# Ensure Group A corresponds to the positive side of your contrast (e.g., "A" or "NLGF")# and Group B corresponds to the negative side (e.g., "B" or "hAPP")if (str_detect(comp_name, "Trisomy vs Disomy")) { label_A <-"Trisomy" label_B <-"Disomy" } elseif (str_detect(comp_name, "NLGF vs.*hAPP")) { label_A <-"NLGF" label_B <-"hAPP" } else {# Fallback just in case a new comparison is added later label_A <-"Group A" label_B <-"Group B" }# 2. Pass the dynamic labels into the GSEA functionrun_gsea_analysis(ranks = ranks,comparison_name = comp_name,geneset_name = gset_name, pathways = gset_pathways, base_objects_dir = objects_dir, base_graphs_dir = graphs_dir, group_A_label = label_A,group_B_label = label_B,color_A = GLOBAL_CONFIG$col_A,color_B = GLOBAL_CONFIG$col_B )gc() # Memory cleanup })})
Single pathway enrichment plot
Code
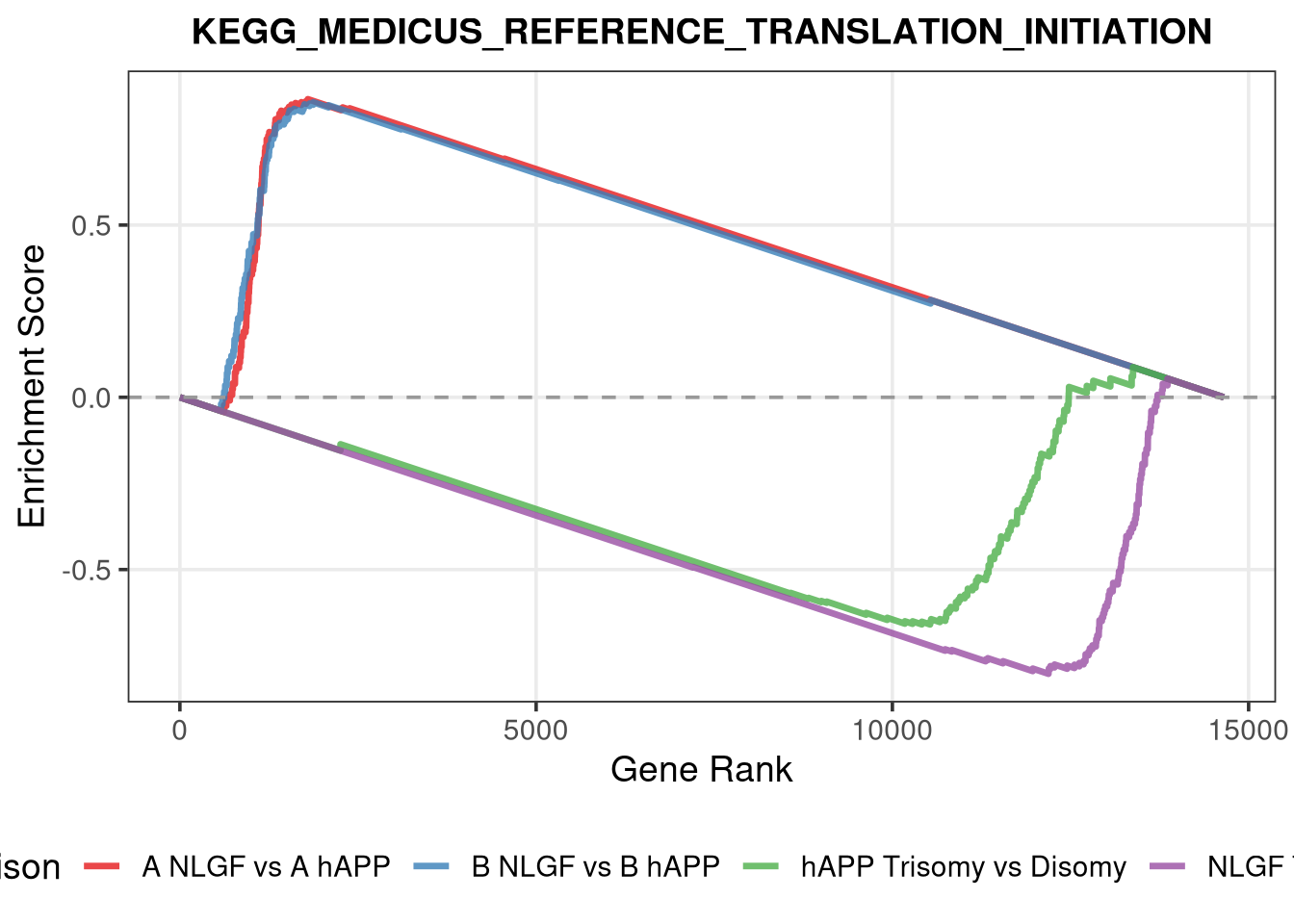
term_to_plot <-"KEGG_MEDICUS_REFERENCE_TRANSLATION_INITIATION"# 1. Flatten your master gene set list so we can find the term easily# (unname prevents "GO_KEGG.TermName" prefixes)all_pathways_flat <-do.call(c, unname(gene_sets_master))# 2. Check if the term exists before loopingif (!term_to_plot %in%names(all_pathways_flat)) {stop(glue("Could not find pathway '{term_to_plot}' in gene_sets_master."))}pathway_genes <- all_pathways_flat[[term_to_plot]]# 3. Create a specific output folderspecific_graphs_dir <-file.path(graphs_dir, "Specific_Pathway_Plots")dir.create(specific_graphs_dir, showWarnings =FALSE, recursive =TRUE)# 4. Loop and Plotpurrr::walk2(comparisons_master, names(comparisons_master), function(ranks, comp_name) {message(glue("Plotting {term_to_plot} for {comp_name}..."))# Generate the GSEA enrichment plot gsea_plot <-plotEnrichment(pathway_genes, ranks) +labs(title = term_to_plot,subtitle =glue("Comparison: {comp_name}"),x ="Rank",y ="Enrichment Score" ) +theme_bw(base_size =14) +theme(plot.title =element_text(face ="bold", hjust =0.5),plot.subtitle =element_text(hjust =0.5, color ="grey40"),panel.grid =element_blank() )# Saveggsave(filename =file.path(specific_graphs_dir, glue("{term_to_plot}_{comp_name}.pdf")),plot = gsea_plot,width =8, height =5 )})
Combined GSEA plots
Define function
Code
plot_combined_gsea <-function(pathway_name, rank_list, pathways_obj, graphs_dir) {# --- A. Sanitize Filename --- safe_pathway_name <-gsub("[[:punct:]]", "_", pathway_name)# --- B. Check if pathway exists ---if (!pathway_name %in%names(pathways_obj)) {warning(glue("Pathway '{pathway_name}' not found in the provided pathway object."))return(NULL) } gene_set <- pathways_obj[[pathway_name]] plot_data_list <-list()# --- C. Loop through comparisons ---for (comp_name innames(rank_list)) { ranks <- rank_list[[comp_name]]# Generate the standard fgsea plot object# Note: We suppress messages to avoid "calibrating stats" spam g <-suppressMessages(plotEnrichment(gene_set, ranks))# Extract the data frame (columns are typically 'rank' and 'ES') df <- g$data df$Comparison <- comp_name plot_data_list[[comp_name]] <- df }# --- D. Combine data --- combined_df <-bind_rows(plot_data_list)# --- E. Plot --- p <-ggplot(combined_df, aes(x = rank, y = ES, color = Comparison)) +geom_line(linewidth =1.2, alpha =0.8) +# Add zero line for referencegeom_hline(yintercept =0, linetype ="dashed", color ="grey60") +labs(title = pathway_name,x ="Gene Rank",y ="Enrichment Score" ) +# Use distinct colors for the different comparisons (hAPP vs NLGF)scale_color_brewer(palette ="Set1") +theme_bw(base_size =14) +theme(legend.position ="bottom",plot.title =element_text(size =14, face ="bold", hjust =0.5),panel.grid.minor =element_blank() )# --- F. Save ---# Create a subfolder for these combined plots combined_out_dir <-file.path(graphs_dir, "Combined_GSEA_Plots")dir.create(combined_out_dir, showWarnings =FALSE, recursive =TRUE) file_name <-file.path(combined_out_dir, glue("{safe_pathway_name}_Combined.pdf"))ggsave(filename = file_name, plot = p, width =8, height =6)return(p)}
Combined enrichment — specific term
Code
term_to_plot <-"KEGG_MEDICUS_REFERENCE_TRANSLATION_INITIATION"# 1. Flatten the master list so we can search all pathways at onceall_pathways_flat <-do.call(c, unname(gene_sets_master))combined_plot <-plot_combined_gsea(pathway_name = term_to_plot,rank_list = comparisons_master, # Uses your hAPP and NLGF rankspathways_obj = all_pathways_flat, # Uses the flattened listgraphs_dir = graphs_dir)print(combined_plot)