Code
suppressPackageStartupMessages({
library(data.table)
library(qs2)
library(dplyr)
library(ggplot2)
library(ggrepel)
library(purrr)
library(stringr)
library(clusterProfiler)
library(org.Mm.eg.db)
library(enrichplot)
library(ggVennDiagram)
})==================================================================
Author: Carlo Zanetti
This script evaluates the consistency between MSstats and limma. It merges results from both methods to identify shared biological signals and pipeline specific differences.
Data loading and visualisation
Import Msstats and limma data
Gene mapping and Annotation
Only necessary for Msstats
Thresholding and categorisation
Apply significance cut-offs (FDR <0.05, |log2FC| > 0.5)
Concordance analysis
Calculate pearson correlation between log2FC values of the 2 pipelines for each comparison
Visualisation
Plots concordance scatter plots between each pipeline and comparison.
Labels top 30 most highly changed genes using ggrepel
Produces tables summarising detction overlap and pipeline-specific abundant proteins
==================================================================
suppressPackageStartupMessages({
library(data.table)
library(qs2)
library(dplyr)
library(ggplot2)
library(ggrepel)
library(purrr)
library(stringr)
library(clusterProfiler)
library(org.Mm.eg.db)
library(enrichplot)
library(ggVennDiagram)
})base_dir <- "/nemo/lab/destrooperb/home/shared/zanettc/millie_proteomics"
run_num <- "run5"
results_dir <- file.path(base_dir, "results", run_num)
objects_dir <- file.path(base_dir, "data", "processed", run_num)
millie_dir <- file.path(base_dir, "data", "processed_millie")
graphs_dir <- file.path(results_dir, "concordance")
millie_f_v_c <- read.csv(file.path(millie_dir, "limma_all_proteins_Plaque_far_vs_Control.csv"))
millie_n_v_c <- read.csv(file.path(millie_dir, "limma_all_proteins_Plaque_near_vs_Control.csv"))
millie_n_v_f <- read.csv(file.path(millie_dir, "limma_all_proteins_Plaque_near_vs_Far.csv"))
res_df <- fread(file.path(results_dir, "Full_GroupComparison_Results.csv"))
clean_spec_raw <- fread(file.path(base_dir, "data/processed/run1/clean_spec_raw.csv"))protein_dictionary <- clean_spec_raw %>%
dplyr::select(Protein = PG.ProteinGroups,
Gene = PG.Genes,
Description = PG.ProteinDescriptions) %>%
distinct()
msstats_ready <- res_df %>%
# Filter for your specific comparison (Label) and handle NAs
filter(!is.na(adj.pvalue), is.finite(log2FC)) %>%
# Join with dictionary
left_join(protein_dictionary, by = "Protein") %>%
# Clean up Gene names and define significance
mutate(
# Fallback to Protein ID if Gene name is missing or empty
Gene = ifelse(is.na(Gene) | Gene == "", Protein, Gene),
# Define significance categories
Status = case_when(
adj.pvalue < 0.05 & log2FC > 0.5 ~ "Upregulated",
adj.pvalue < 0.05 & log2FC < -0.5 ~ "Downregulated",
TRUE ~ "Not Significant"
)
) %>%
# Ranking for plotting/reporting
group_by(Label, Status) %>%
mutate(Rank = rank(adj.pvalue, ties.method = "first")) %>%
ungroup()
msstats_ready %>%
filter(Label == "PlaqueNear_vs_PlaqueFar") %>%
filter(log2FC > 0.5 & adj.pvalue < 0.05)# Add labels to Millie's data to match MSstats 'Label' names
# Check your msstats_ready$Label to ensure these strings match exactly!
millie_combined <- bind_rows(
millie_f_v_c %>% mutate(Label = "PlaqueFar_vs_Control"),
millie_n_v_c %>% mutate(Label = "PlaqueNear_vs_Control"),
millie_n_v_f %>% mutate(Label = "PlaqueNear_vs_PlaqueFar")
) %>%
dplyr::rename(
log2FC_limma = logFC,
adj.pvalue_limma = adj.P.Val
) %>%
mutate(
Status_limma = case_when(
adj.pvalue_limma < 0.05 & log2FC_limma > 0.5 ~ "Upregulated",
adj.pvalue_limma < 0.05 & log2FC_limma < -0.5 ~ "Downregulated",
TRUE ~ "Not Significant"
)
) %>%
dplyr::select(Gene, Label, log2FC_limma, adj.pvalue_limma, Status_limma)
millie_combinedwrite.csv(millie_combined, file.path(millie_dir, "millie_combined.csv"))comparison_df <- msstats_ready %>%
dplyr::select(Protein, Gene, Label, log2FC_msstats = log2FC, adj.pvalue_msstats = adj.pvalue, Status_msstats = Status) %>%
full_join(millie_combined, by = c("Gene", "Label")) %>%
mutate(
# Create a column to easily find discrepancies
Agreement = case_when(
Status_msstats == Status_limma ~ "Consistent",
Status_msstats == "Not Significant" | Status_limma == "Not Significant" ~ "Pipeline Specific",
TRUE ~ "Contradictory" # One says Up, one says Down
)
)
# Preview the overlap
table(comparison_df$Status_msstats, comparison_df$Status_limma)
Downregulated Not Significant Upregulated
Downregulated 3 15 0
Not Significant 25 15587 33
Upregulated 0 645 177comparison_df %>% filter(Gene== "Cd44")plot_data <- comparison_df %>%
# 1. Determine Significance categories based on your previous Status columns
mutate(
Significance = case_when(
Status_msstats != "Not Significant" & Status_limma != "Not Significant" ~ "Significant in Both",
Status_msstats != "Not Significant" & Status_limma == "Not Significant" ~ "Significant in MSstats only",
Status_msstats == "Not Significant" & Status_limma != "Not Significant" ~ "Significant in limma only",
TRUE ~ "Not Significant"
),
# 2. Calculate distance from origin (Magnitude in both) to pick top labels
# Using coalesce to handle NAs just in case a protein is only in one pipeline
dist = sqrt(coalesce(log2FC_msstats, 0)^2 + coalesce(log2FC_limma, 0)^2)
)
# Define your colors to match the exact spelling of categories
sig_colors <- c(
"Significant in Both" = "purple",
"Significant in MSstats only" = "#00BFC4", # Cyan/Blue
"Significant in limma only" = "#F8766D", # Coral/Red
"Not Significant" = "grey80"
)
# --- STEP 2: Loop through each comparison and plot ---
comparisons <- unique(plot_data$Label)
for (comp in comparisons) {
cat("Plotting:", comp, "\n")
# Subset data for the current comparison
df_sub <- plot_data %>% filter(Label == comp)
# Check if we have data to plot before proceeding
if(nrow(df_sub %>% filter(!is.na(log2FC_msstats) & !is.na(log2FC_limma))) == 0) {
message(paste("No overlapping data found for:", comp, "- skipping."))
next
}
# Get top 30 genes to label based on distance from origin
genes_to_label <- df_sub %>%
filter(Significance != "Not Significant") %>%
slice_max(order_by = dist, n = 30, with_ties = FALSE)
# Calculate Pearson correlation for the subtitle
cor_val <- round(cor(df_sub$log2FC_msstats, df_sub$log2FC_limma, use = "complete.obs"), 3)
# Clean up the comparison name for the plot title (e.g., "PlaqueNear_vs_Control" -> "Plaque Near vs Control")
clean_title <- gsub("_", " ", comp)
# Optional: Add spaces before capital letters if your label is CamelCase
# clean_title <- gsub("([a-z])([A-Z])", "\\1 \\2", clean_title)
#### settings to make plot look pretty -> so all genes fit nicely in plot
x_bulk <- quantile(df_sub$log2FC_limma, probs = c(0.01, 0.99), na.rm = TRUE)
y_bulk <- quantile(df_sub$log2FC_msstats, probs = c(0.01, 0.99), na.rm = TRUE)
#Get the range of the proteins we actually want to label
x_label_range <- range(genes_to_label$log2FC_limma, na.rm = TRUE)
y_label_range <- range(genes_to_label$log2FC_msstats, na.rm = TRUE)
# 3. Final Limits: Use the wider of the two ranges so labels aren't cut off
final_xlim <- c(min(x_bulk[1], x_label_range[1]), max(x_bulk[2], x_label_range[2]))
final_ylim <- c(min(y_bulk[1], y_label_range[1]), max(y_bulk[2], y_label_range[2]))
# Add 15% padding so labels have room to "repel" into
x_pad <- diff(final_xlim) * 0.15
y_pad <- diff(final_ylim) * 0.15
# Build The Plot
p <- ggplot(df_sub, aes(x = log2FC_limma, y = log2FC_msstats)) +
# 1. Diagonal and Axes
geom_abline(slope = 1, intercept = 0, color = "red", linetype = "solid", linewidth = 0.5) +
geom_hline(yintercept = 0, linetype = "dashed", color = "grey50") +
geom_vline(xintercept = 0, linetype = "dashed", color = "grey50") +
# 2. Scatter Points
geom_point(aes(color = Significance), alpha = 0.6, size = 2) +
# 3. Optimized Labels (Mapped to your "Gene" column)
geom_label_repel(
data = genes_to_label,
aes(label = Gene, color = Significance),
size = 4,
fontface = "bold",
box.padding = 0.5,
point.padding = 0.3,
force = 2,
max.overlaps = Inf,
min.segment.length = 0,
show.legend = FALSE,
fill = "white"
) +
# 4. Styling
scale_color_manual(values = sig_colors) +
coord_cartesian(
xlim = c(final_xlim[1] - x_pad, final_xlim[2] + x_pad),
ylim = c(final_ylim[1] - y_pad, final_ylim[2] + y_pad),
expand = FALSE # Keeps our manual padding exact
) +
theme_bw(base_size = 16) +
labs(
title = paste("Concordance:", clean_title),
subtitle = paste("limma vs. MSstats | Pearson r =", cor_val),
x = "limma Log2FC",
y = "MSstats Log2FC",
color = "Significance (FDR < 0.05)"
) +
theme(
legend.position = "bottom",
legend.text = element_text(size = 12),
legend.title = element_text(size = 12, face = "bold"),
panel.grid.minor = element_blank(),
plot.title = element_text(face = "bold", size = 18),
axis.title = element_text(face = "bold")
) +
guides(color = guide_legend(
override.aes = list(size = 5),
nrow = 2
))
# Print to RStudio viewer/console so you can see them as they generate
print(p)
# Sanitize filename (replace spaces/special chars with underscores)
safe_filename <- str_replace_all(comp, "[^A-Za-z0-9]+", "_")
safe_filename <- str_replace(safe_filename, "_$", "")
# Save to PDF
ggsave(
filename = file.path(graphs_dir, paste0("MSstats_vs_limma_concordance_", safe_filename, ".pdf")),
plot = p,
width = 10,
height = 10,
dpi = 300
)
}Plotting: PlaqueNear_vs_Control Warning: Removed 222 rows containing missing values or values outside the scale range
(`geom_point()`).
Removed 222 rows containing missing values or values outside the scale range
(`geom_point()`).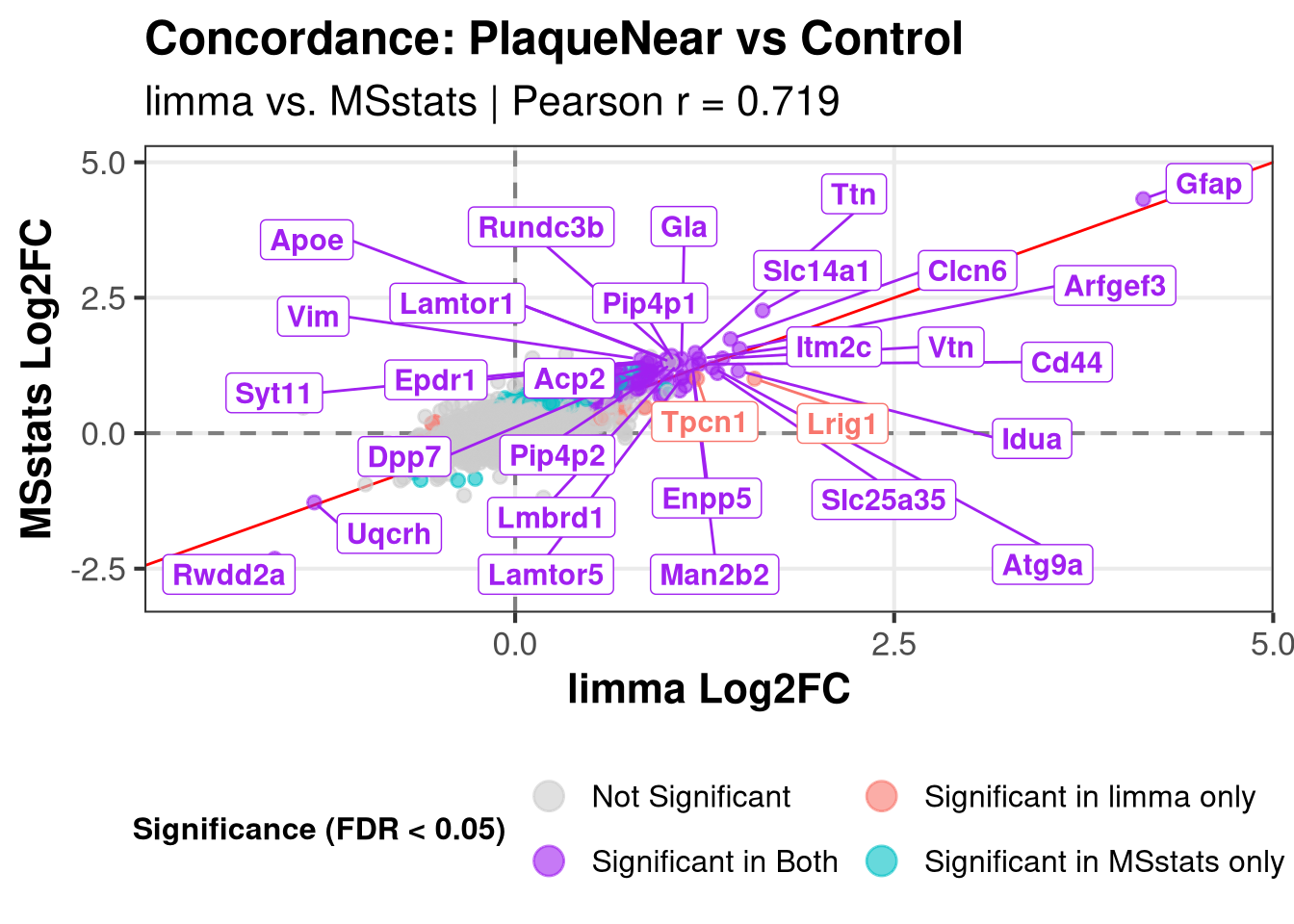
Plotting: PlaqueFar_vs_Control Warning: Removed 222 rows containing missing values or values outside the scale range
(`geom_point()`).
Removed 222 rows containing missing values or values outside the scale range
(`geom_point()`).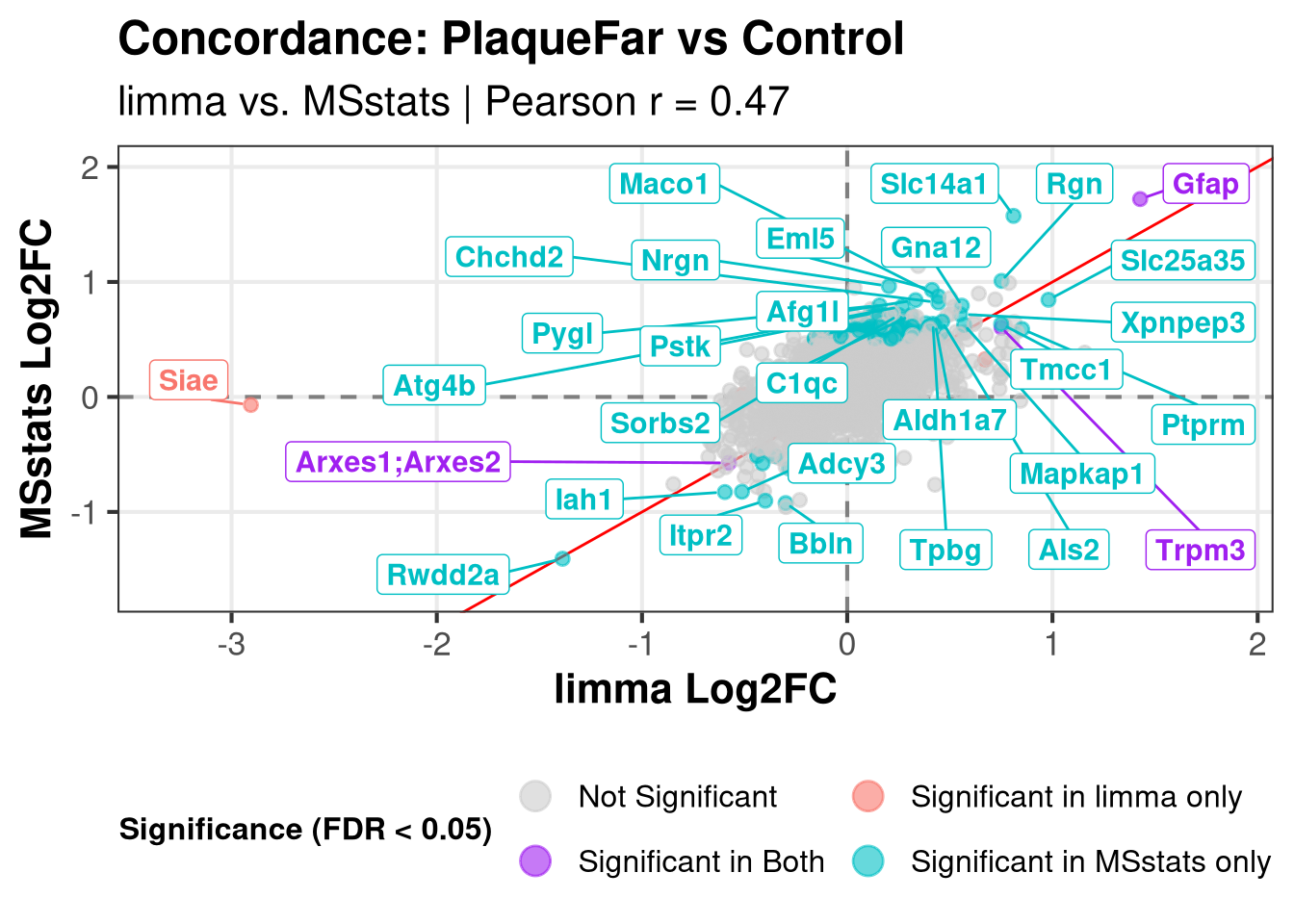
Plotting: PlaqueNear_vs_PlaqueFar Warning: Removed 221 rows containing missing values or values outside the scale range
(`geom_point()`).Warning: Removed 221 rows containing missing values or values outside the scale range
(`geom_point()`).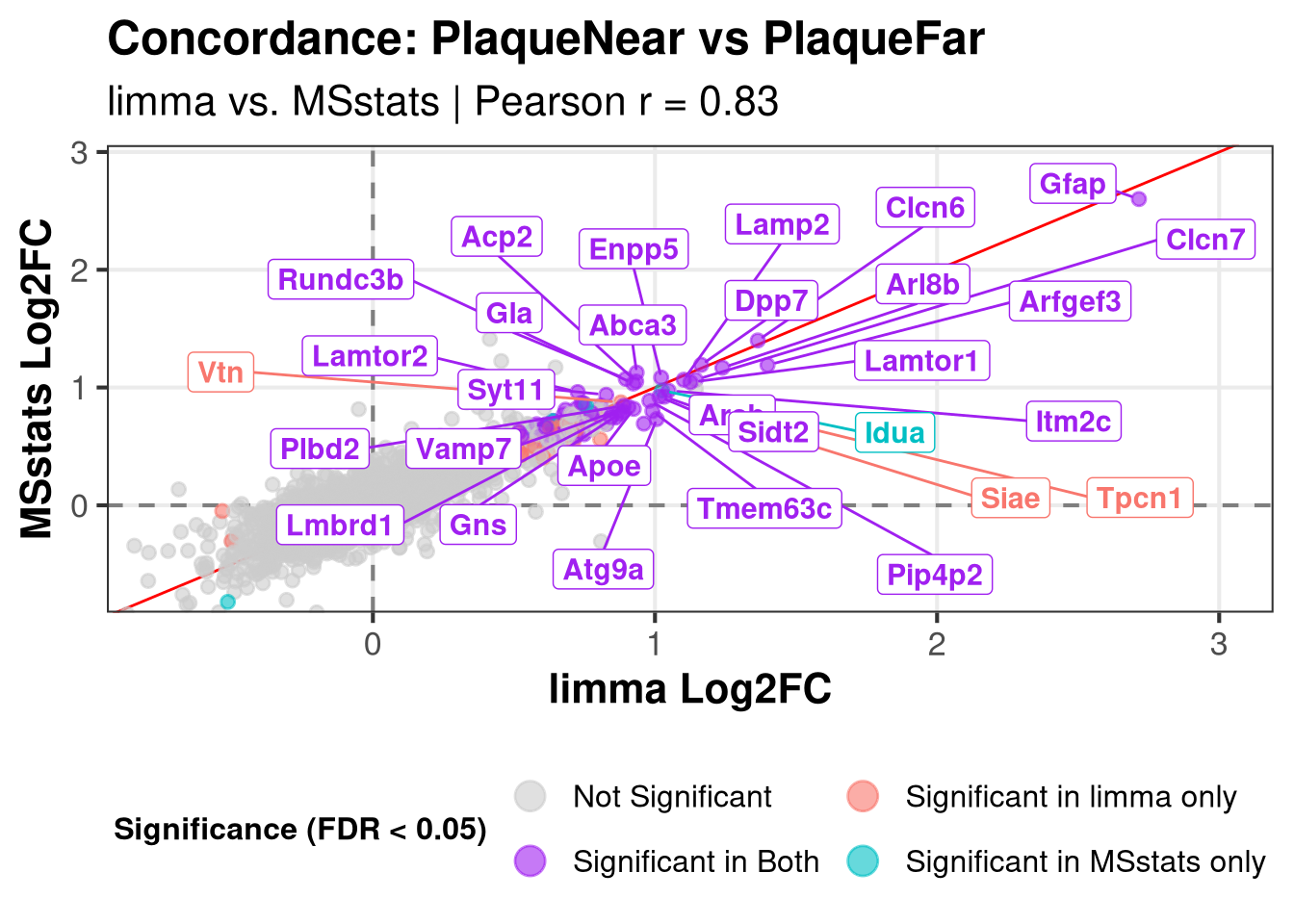
cat("All plots generated and saved successfully to:", graphs_dir, "\n")All plots generated and saved successfully to: /nemo/lab/destrooperb/home/shared/zanettc/millie_proteomics/results/run5/concordance Summarises the overlap in detection and significance.
summary_table <- comparison_df %>%
group_by(Label) %>%
summarise(
# 1. Total Detected overall (union of both methods)
Total_Detected_Overall = n(),
# 2. Detected by specific methods
Detected_MSstats = sum(!is.na(log2FC_msstats)),
Detected_limma = sum(!is.na(log2FC_limma)),
# 3. Both Detected (Shared column)
Both_Detected = sum(!is.na(log2FC_msstats) & !is.na(log2FC_limma)),
# 4. Significant (FDR < 0.05)
Sig_MSstats = sum(Status_msstats != "Not Significant", na.rm = TRUE),
Sig_limma = sum(Status_limma != "Not Significant", na.rm = TRUE),
# 5. Both Significant (Shared column)
Both_Significant = sum(Status_msstats != "Not Significant" &
Status_limma != "Not Significant", na.rm = TRUE)
) %>%
# Clean up the Label for readability
mutate(Label = gsub("_", " ", Label))
summary_table#Filter for signficant msstats proteins and check if detected by Millie
msstats_sig_analysis <- comparison_df %>%
filter(Status_msstats != "Not Significant") %>%
mutate(
Detected_by_Millie = ifelse(!is.na(log2FC_limma), "Yes", "No")
)
# Summarize the findings per comparison
detection_overlap <- msstats_sig_analysis %>%
group_by(Label, Detected_by_Millie) %>%
tally(name = "Protein_Count") %>%
tidyr::pivot_wider(names_from = Detected_by_Millie, values_from = Protein_Count, names_prefix = "Detected_by_limma_")
print(detection_overlap)# A tibble: 3 × 3
# Groups: Label [3]
Label Detected_by_limma_No Detected_by_limma_Yes
<chr> <int> <int>
1 PlaqueFar_vs_Control 3 310
2 PlaqueNear_vs_Control 4 455
3 PlaqueNear_vs_PlaqueFar NA 74limma_exclusive_sig <- comparison_df %>%
filter(Label == "PlaqueNear_vs_PlaqueFar") %>%
filter(Status_limma != "Not Significant" & Status_msstats == "Not Significant") %>%
dplyr::select(Gene,
log2FC_limma, adj.pvalue_limma,
log2FC_msstats, adj.pvalue_msstats) %>%
arrange(adj.pvalue_limma)
limma_exclusive_sigmsstats_exclusive_sig <- comparison_df %>%
filter(Label == "PlaqueNear_vs_PlaqueFar") %>%
filter(Status_limma == "Not Significant" & Status_msstats != "Not Significant") %>%
dplyr::select(Gene,
log2FC_limma, adj.pvalue_limma,
log2FC_msstats, adj.pvalue_msstats) %>%
arrange(adj.pvalue_limma)
msstats_exclusive_sigcheck_cutoffs_msstats <- comparison_df %>%
filter(Label == "PlaqueNear_vs_PlaqueFar") %>%
filter(adj.pvalue_msstats < 0.05 & Status_msstats == "Not Significant") %>%
dplyr::select(Gene, log2FC_msstats, adj.pvalue_msstats)
check_cutoffs_msstatscheck_cutoffs_limma <- comparison_df %>%
filter(Label == "PlaqueNear_vs_PlaqueFar") %>%
filter(adj.pvalue_msstats < 0.05 & Status_limma == "Not Significant") %>%
dplyr::select(Gene, log2FC_limma, adj.pvalue_limma, log2FC_msstats, adj.pvalue_msstats)
check_cutoffs_limma# 1. Isolate significant genes for Near vs Control (MSstats)
near_vs_control_sig <- comparison_df %>%
filter(Label == "PlaqueNear_vs_Control" & Status_msstats != "Not Significant") %>%
pull(Gene) %>%
unique()
# 2. Isolate significant genes for Far vs Control (MSstats)
far_vs_control_sig <- comparison_df %>%
filter(Label == "PlaqueFar_vs_Control" & Status_msstats != "Not Significant") %>%
pull(Gene) %>%
unique()
# 3. Calculate intersections and differences
shared_msstats_genes <- intersect(near_vs_control_sig, far_vs_control_sig)
unique_to_near <- setdiff(near_vs_control_sig, far_vs_control_sig)
unique_to_far <- setdiff(far_vs_control_sig, near_vs_control_sig)
cat("MSstats Significance Overlap Summary:\n")MSstats Significance Overlap Summary:cat("-------------------------------------\n")-------------------------------------cat("Total Significant in Near vs Control:", length(near_vs_control_sig), "\n")Total Significant in Near vs Control: 459 cat("Total Significant in Far vs Control:", length(far_vs_control_sig), "\n")Total Significant in Far vs Control: 313 cat("Shared between both (Intersection):", length(shared_msstats_genes), "\n")Shared between both (Intersection): 137 cat("Unique to Near vs Control:", length(unique_to_near), "\n")Unique to Near vs Control: 322 cat("Unique to Far vs Control:", length(unique_to_far), "\n\n")Unique to Far vs Control: 176 # 1. Group the significant gene lists into a named list
msstats_venn_list <- list(
"Near vs Control" = near_vs_control_sig,
"Far vs Control" = far_vs_control_sig
)
# 2. Generate the Venn Diagram
venn_plot <- ggVennDiagram(msstats_venn_list,
label_alpha = 0,
set_color = "black",
label = "both") + # Shows count and percentage
scale_fill_gradient(low = "white", high = "#00BFC4") +
theme_void(base_size = 14) +
labs(title = "MSstats: Significant Proteins Overlap", fill = "Gene Count") +
theme(
plot.title = element_text(face = "bold", hjust = 0.5),
# Add large margins (Top, Right, Bottom, Left) to prevent label clipping
plot.margin = margin(20, 60, 20, 60)
) +
# This allows labels to draw outside the "official" plot area
coord_cartesian(clip = "off")Coordinate system already present.
ℹ Adding new coordinate system, which will replace the existing one.# Print the plot
print(venn_plot)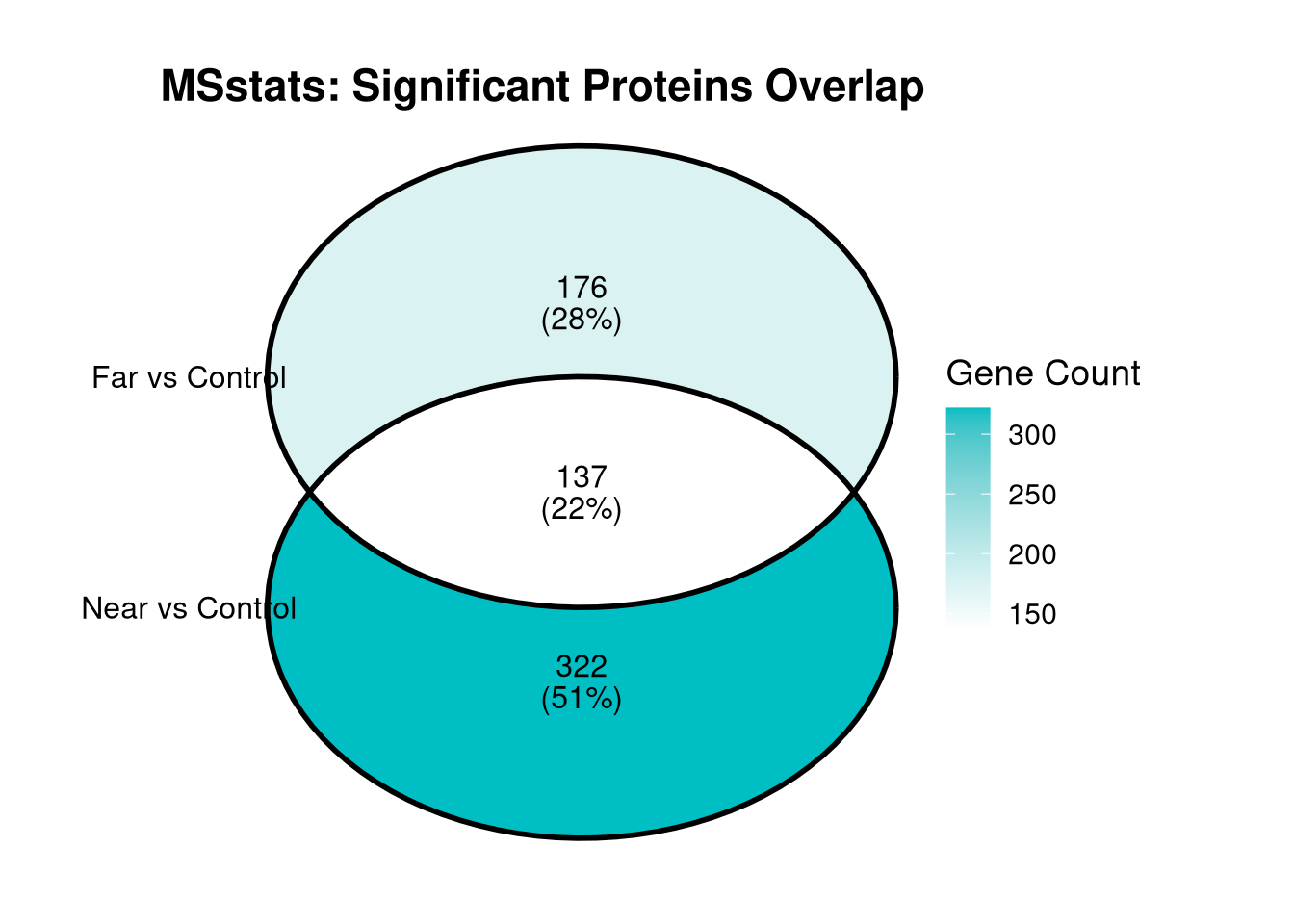
# 4. Save the plot to your graphs directory
ggsave(
filename = file.path(graphs_dir, "MSstats_Near_vs_Far_Venn.pdf"),
plot = venn_plot,
width = 8,
height = 6,
dpi = 300
)msstats_far_only_df <- comparison_df %>%
filter(Label == "PlaqueFar_vs_Control") %>%
filter(Gene %in% unique_to_far) %>%
dplyr::select(Gene, log2FC_msstats, adj.pvalue_msstats, Status_msstats) %>%
arrange(adj.pvalue_msstats)
# 2. Preview the top of the list
cat("Top 10 proteins unique to Plaque Far vs Control (MSstats):\n")Top 10 proteins unique to Plaque Far vs Control (MSstats):msstats_far_only_dffwrite(msstats_far_only_df, file.path(results_dir, "msstats_plaque_far_vs_control_only.csv"))