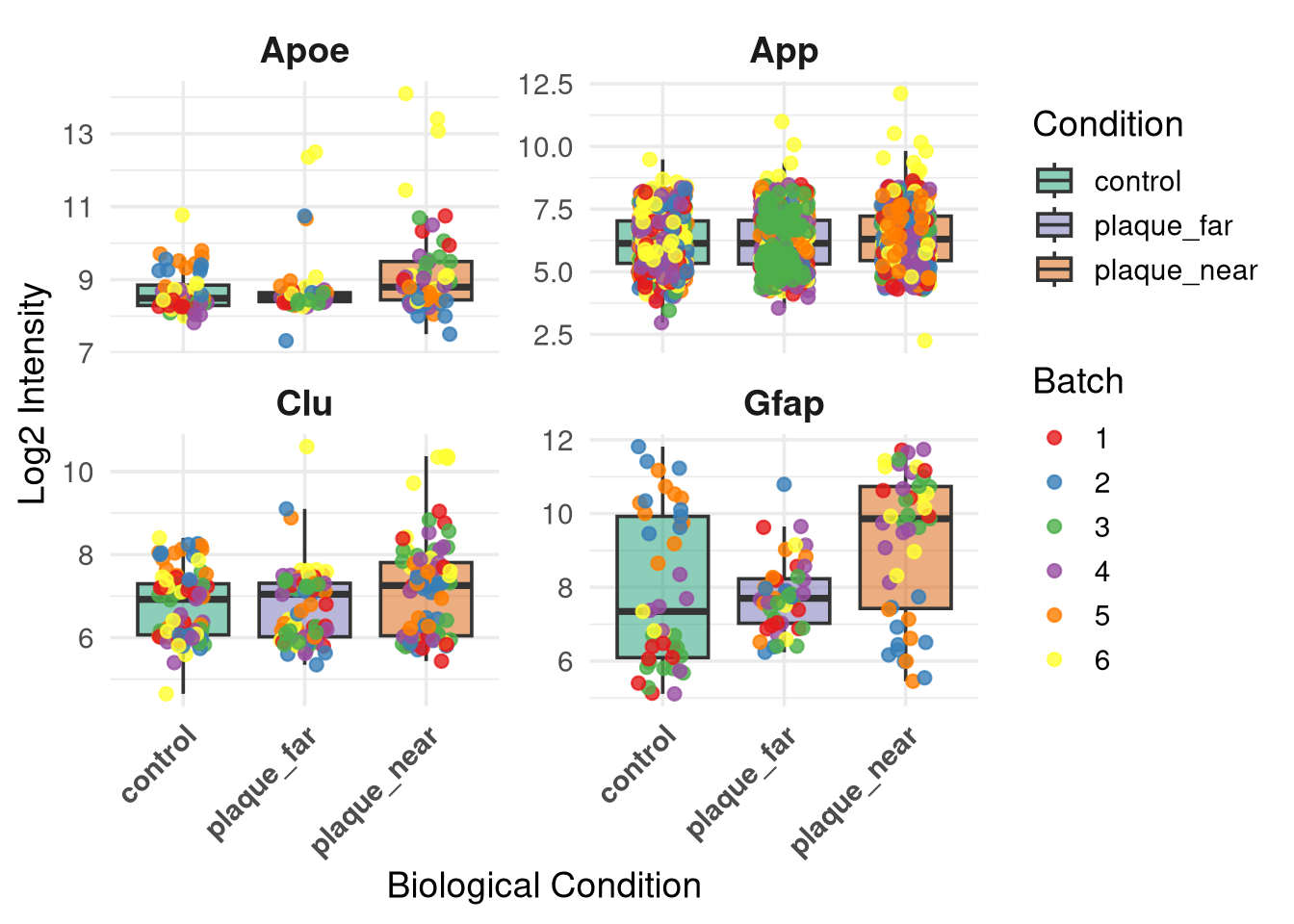
base_dir <-"/nemo/lab/destrooperb/home/shared/zanettc/millie_proteomics"run_num <-"run1"# Pointing to where you saved the clean data, and where to output the plotobjects_dir <-file.path(base_dir, "data", "processed", run_num)results_dir <-file.path(base_dir, "results", run_num)
Warning: Removed 672 rows containing non-finite outside the scale range
(`stat_boxplot()`).
Removed 672 rows containing missing values or values outside the scale range
(`geom_point()`).
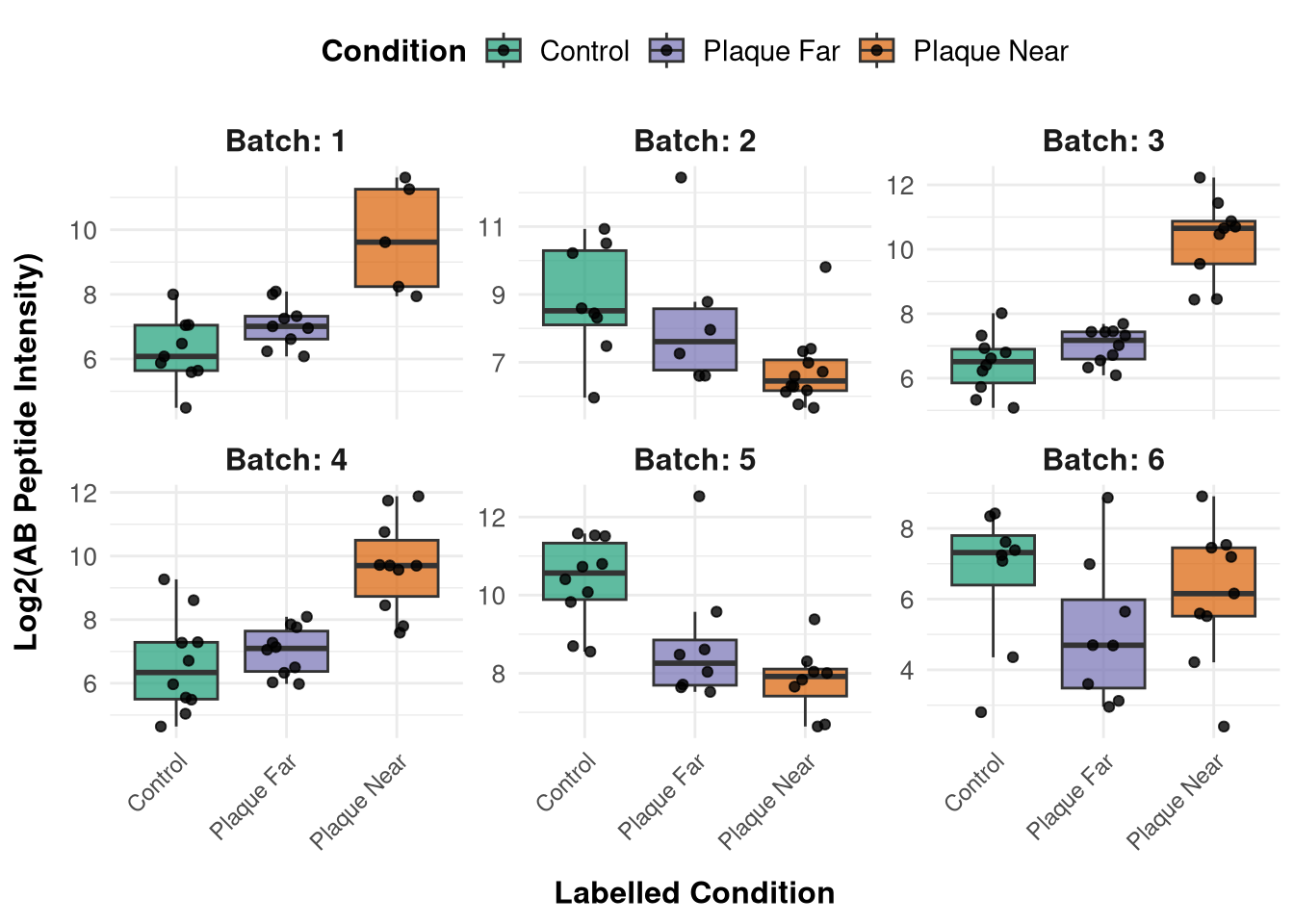
Pull in MSstats data to display Aß tryptic peptide plot